
Articles
- Page Path
- HOME > Acute Crit Care > Volume 30(1); 2015 > Article
- Case Report Hemophagocytic Lymphohistiocytosis after Lung Transplantation
- Ah Young Leem, M.D., Sung Woo Moon, M.D., Song Yee Kim, M.D., Moo Suk Park, M.D., Young Sam Kim, M.D., Se Kyu Kim, M.D., Joon Chang, M.D., Hyo Chae Paik, M.D.*, June Won Cheong, M.D.†, Kyung Soo Chung, M.D.
-
Korean Journal of Critical Care Medicine 2015;30(1):38-41.
DOI: https://doi.org/10.4266/kjccm.2015.30.1.38
Published online: February 28, 2015
Division of Pulmonology, Department of Internal Medicine, Institute of Chest Disease, Severance Hospital, Yonsei University College of Medicine, Seoul, Korea
*Division of Pulmonology, Department of Thoracic and Cardiovascular Surgery, Institute of Chest Disease, Severance Hospital, Yonsei University College of Medicine, Seoul, Korea.
��Division of Hematology, Department of Internal Medicine, Institute of Chest Disease, Severance Hospital, Yonsei University College of Medicine, Seoul, Korea.
- Correspondence to: Kyung Soo Chung, Division of Pulmonology, Department of Internal Medicine, Institute of Chest Disease, Severance Hospital, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 120-752, Korea Tel: +82-2-2227-4203, Fax: +82- 2-393-6884 E-mail: chungks@yuhs.ac
Copyright © 2015 The Korean Society of Critical Care Medicine
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 57,913 Views
- 80 Download
Abstract
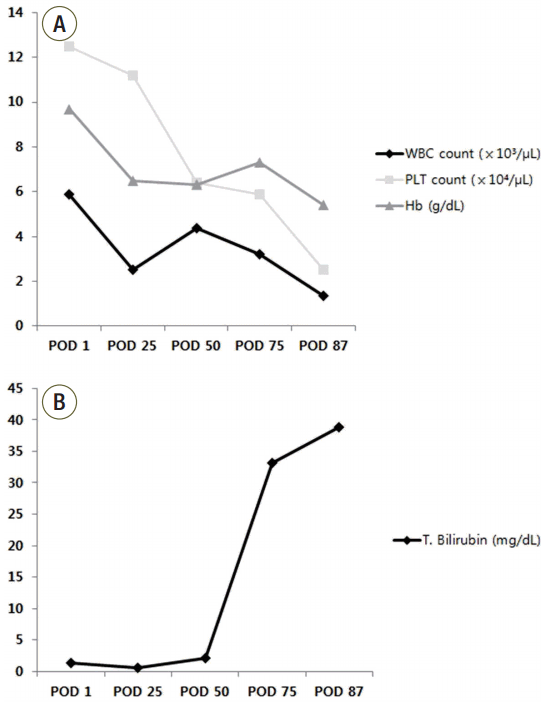
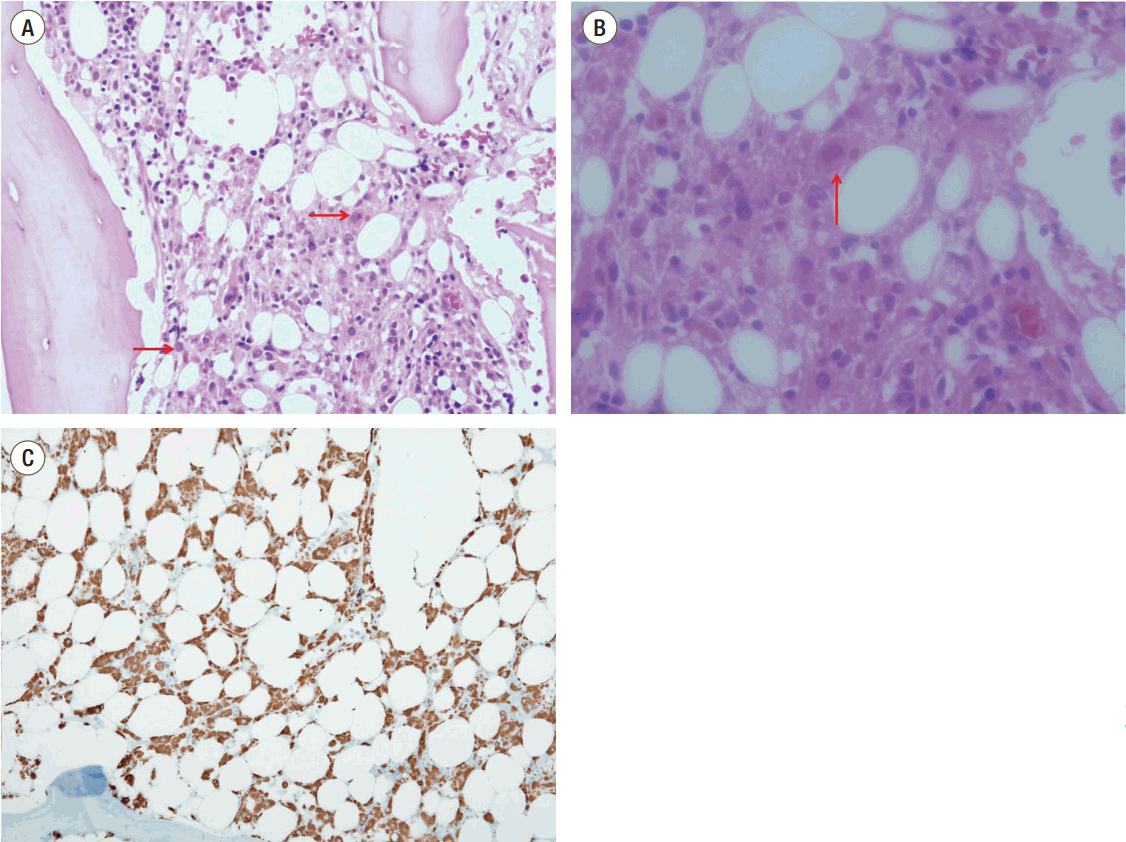
- Hemophagocytic lymphohistiocytosis (HLH) is a rare but fatal complication after solid organ transplantation. Acquired forms of HLH are described in association with severe sepsis, autoimmune disorders, malignancy, immune-compromised states, infections, and solid organ transplantation. We experienced a case of hemophagocytic lymphohistiocytosis after bilateral lung transplantation. Leukopenia, thrombocytopenia, and hyperbilirubinemia were noted and became aggravated 50 days after transplantation. Diagnosis of HLH was based on clinical and laboratory findings of splenomegaly, cytopenia, elevated ferritin, elevated interleukin-2 receptor, and hemophagocytosis in bone marrow. Other features such as elevated bilirubin, lactate dehydrogenase, and D-dimer which can be present in HLH were also noted. The patient was immediately treated with etoposide and dexamethasone. Despite aggressive therapy, the patient deteriorated and died. Awareness of the diagnostic criteria of HLH after lung transplantation is important for clinicians.
Case Report
Discussion
- 1. Janka GE, Lehmberg K. Hemophagocytic syndromes an update. Blood Rev 2014;28:135-42.ArticlePubMed
- 2. Oto T, Snell GI, Goto K, Miyoshi S. Hemophagocytic syndrome: a rare but specific complication of lung transplantation. J Thorac Cardiovasc Surg 2010;140:e58-9.ArticlePubMed
- 3. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124-31.ArticlePubMed
- 4. Diaz-Guzman E, Dong B, Hobbs SB, Kesler MV, Hayes D Jr. Hemophagocytic lymphohistiocytosis after lung transplant: report of 2 cases and a literature review. Exp Clin Transplant 2011;9:217-22.PubMed
- 5. Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH Jr, Simmons RL, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 1979;44:993-1002.ArticlePubMed
- 6. Karras A, Thervet E, Legendre C, Groupe Coopératif de transplantation d’lle de France. Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation 2004;77:238-43.ArticlePubMed
 PubReader
PubReader ePub Link
ePub Link Cite
Cite


