Abstract
- Severe hyperammonemia can occur as a result of inherited or acquired liver enzyme defects in the urea cycle, among which ornithine transcarbamylase deficiency (OTCD) is the most common form. We report a very rare case of a 45-year-old Korean male who was admitted to the intensive care unit (ICU) due to severe septic shock with acute respiratory failure caused by Pneumocystis jiroveci pneumonia. During his ICU stay with ventilator care, the patient suffered from marked hyperammonemia (>1,700 μg/dL) with abrupt mental change leading to life-threatening cerebral edema. Despite every effort including continuous renal replacement therapy and use of a molecular adsorbent recirculating system (extracorporeal liver support–albumin dialysis) to lower his serum ammonia level, the patient was not recovered. The lethal hyperammonemia in the patient was later proven to be a manifestation of acquired liver enzyme defect known as OTCD, which is triggered by serious catabolic conditions, such as severe septic shock with acute respiratory failure.
-
Keywords: cerebral edema; hyperammonemia; ornithine transcarbamylase deficiency; respiratory failure; septic shock
Patients with acute hyperammonemia are frequently encountered in the intensive care unit (ICU) and are known to have serious morbidity and mortality. If the arterial ammonia level exceeds 200 μmol/L, cerebral edema and brain herniation can occur as brain astrocytes rapidly metabolize ammonia to glutamine, with a subsequent rise in intracellular osmolarity leading to astrocyte swelling and loss.[1-3]
The causes of hyperammonemia can be divided into two main processes that either increase ammonia production or decrease ammonia elimination. Conditions that increase ammonia production include infection and protein load or increased catabolism such as severe exercise, seizure, trauma, starvation, gastrointestinal (GI) hemorrhage, steroid treatment, and chemotherapy, while conditions associated with decreased ammonia elimination comprise acute or chronic liver failure, portosystemic shunt, and various kinds of drugs. Among these conditions, fulminant hepatic failure is the most common cause of acute hyperammonemia in adult ICUs.[4]
However, if the above-mentioned causes can be eliminated and the etiology of hyperammonemia continues to remain elusive with serum ammonia levels disproportionate to the degree of liver function, clinicians should suspect an inborn error of metabolism: urea cycle defect (UCD). As ammonia is metabolized to urea through the urea cycle by several enzymes located mainly in the liver, inborn defects of such enzymes can bring about severe, fatal hyperammonemia even in adulthood, among which ornithine transcarbamylase deficiency (OTCD) is most commonly diagnosed in adults. [4-5]
We report a very rare case of a 45-year-old Korean male who developed lethal hyperammonemia with life-threatening cerebral edema resulting from adult-onset OTCD, which was triggered by severe septic shock with acute respiratory failure due to Pneumocystis jiroveci pneumonia during an ICU stay.
Case Report
A 45-year-old Korean male was admitted to our emergency room (ER) with dyspnea aggravation from 1 day prior. He had complained of coughing with dyspnea for 3 weeks. On arrival, his vital signs showed a mild fever of up to 37.3°C, tachycardia (142 beats/min), and peripheral blood O2 saturation (SpO2) of 88% in room air. His past medical history included liver cirrhosis and chronic hepatitis B from 6 years prior. About 1.5 years prior, he was diagnosed with hepatocellular carcinoma (HCC) and received trans-catheter arterial chemoembolization (TACE) and radiation therapy. After that, his liver function deteriorated continuously, leading to hepatic failure, and he underwent liver transplantation in China about 1 year prior. However, 7 months prior, regular follow-up imaging showed recurrence of HCC and multiple seeding nodules in the abdomen. Thereafter, five additional TACEs were performed with continuous administration of immunosuppressants.
At ER admission, chest imaging revealed patch consolidations, ground glass opacities, and interlobular septal thickening in both lungs, with bilateral pleural effusion (Fig. 1). His initial laboratory exam showed neutrophil counts of 570/μL and slightly elevated C-reactive protein (15.7 mg/dL) and serum creatinine levels (1.56 mg/dL) on a normal liver function test (Table 1). He was diagnosed with pneumonia in a neutropenic state. Despite full coverage of proper antibiotics, his condition worsened steadily, eventually leading to acute respiratory failure and septic shock, which required ventilator care and hemodynamic monitoring in the ICU (hospital day 10).
Bronchial washing cytology with Gomori methenamine silver staining confirmed the diagnosis of Pneumocystis jiroveci pneumonia. Furthermore, blood cultures proved to be positive for G(-) rods and G(+) cocci (hospital day 24). In the meantime, he additionally experienced recurrent duodenal ulcer bleeding with an impending perforation, and endoscopic hemostasis was carried out repeatedly. With the passage of time, his general status was constantly aggravated, and every effort to wean him off of treatment was not successful. At hospital day 41, he underwent abrupt mental change and showed seizure-like movements. On laboratory examination, his ammonia level was measured as 411 μg/dL, despite normal liver function test results. Moreover, blood urea nitrogen and creatinine levels rose up to 72.2 mg/dL and 3.02 mg/dL, respectively (Table 1). Further neurological exams including electroencephalography diagnosed his condition as metabolic encephalopathy with negative myoclonus. After intravenous phenytoin and midazolam infusion, the seizure-like motions subsided.
At hospital day 43, further exams, such as Doppler ultrasonography, were conducted to evaluate transplanted liver graft function, which displayed normal findings in the liver parenchyma, bile duct, and portal and hepatic vein, including normal Doppler findings. Therefore, under presumptive diagnosis of UCD as a cause of the markedly elevated hyperammonemia, we performed a serum amino acid assay and urine analysis in order to detect urea cycle metabolites (at hospital day 44). Meanwhile, in spite of frequent lactulose enema, administration of oral sodium phenylbutyrate, and continuous renal replacement therapy (CRRT), his ammonia level continually increased to more than 1,700 μg/dL, and his mental status deteriorated even more than before (hospital day 44; Table 1).
Contrast brain magnetic resonance imaging revealed severe swelling of the brain and increased cortical and subcortical gray matter signals suggesting severe hypoxic damage with an impending state of tonsillar and trans-tentorial herniation (Fig. 2). With his family’s consent, a molecular adsorbent recirculating system (MARS, extracorporeal liver support–albumin dialysis) was applied twice to lower the serum ammonia level (hospital day 44, 45). Then, though the serum ammonia level decreased to 956 μg/dL, his mental status did not recover, and the serum ammonia level began to rise again. At hospital day 48, his vital signs became unstable, and metabolic acidosis developed. Finally, he expired due to refractory shock at hospital day 52. Afterwards, markedly elevated excretion of orotic acid was found on a previously performed urine collection analysis, which was a diagnostic finding of ornithine transcarbamylase deficiency (OTCD; Fig. 3).
Discussion
OTCD is the most common inherited disorder among UCDs. The ornithine transcarbamylase (OTC) gene is located on the short arm of the X chromosome and is mainly expressed in the liver.[6-8] Patients with OTCD experience repeated hyperammonemic episodes, resulting in eventual central nervous system (CNS) damage. It is a rare disease with an incidence of about one in every 40,000–80,000 births.[9] As OTCD is an X chromosome–linked disorder, clinical severity differs according to sex. While hemizygous males are usually severely affected during the neonatal period, heterozygous females exhibit various enzymatic activities and clinical courses, ranging from neonatal-onset fatal disease to the absence of symptoms during their lifetime. [10] However, even those who remain asymptomatic into their adult life are able to undergo occasional expressions of symptoms when precipitated by factors such as high protein intake, infection, trauma, surgery, childbirth, or other physiological catabolic stresses.[11] In Korea, adult-onset OTCD is known to be very rare compared to child-onset diseases.
In the present case report, the patient had a previously unhealthy liver with chronic hepatitis B, liver cirrhosis, and HCC. He might have also had recurrent episodes of hepatic encephalopathy resulting from mild hyperammonemia due to his decompensated liver function, though we could not identify relevant data in his medical chart. However, he had gone through a normal growth process, not experiencing seizures or other neurologic disorders before chronic liver disease developed. Additionally, he underwent successful liver transplantation without any complication or graft failure. Even after HCC recurred in his transplanted graft, liver function was constantly normal, and he managed to get along without any problems until he suffered from Pneumocystis jiroveci pneumonia. In addition, we confirmed through Doppler ultrasonography that his transplanted liver did not have any graft dysfunction or anatomical problem in the vessels or ducts except for the recurred HCCs in the parenchyma at the time of abrupt mental change in the ICU. Therefore, considering the disproportionately high levels of serum ammonia with life-threatening cerebral edema and all of the other evidence mentioned above, we suspected UCD as a cause of his hyperammonemia and further confirmed the diagnosis of OTCD based on the high urinary orotic acid levels. To the best of our knowledge, this is the first Korean case of acquired OTCD after liver transplantation, presenting as deadly hyperammonemia in the ICU.
In OTCD, plasma glutamine and alanine levels increase whereas citrulline decreases, resulting from the shortage of the second-passage enzyme ornithine transcarbamylase.[5] A markedly increased level of urine orotic acid is diagnostic of OTCD as in this case.
Symptoms of late-onset OTCD varies from nausea, mild irritability, and protein avoidance to more serious forms such as neuropsychiatric changes, seizures, coma, and even death, which are precipitated by episodes of physiologic stress or catabolism that can lead to hyperammonemia.[11] In this case, the patient experienced Pneumocystis jiroveci pneumonia and consecutive bacterial septic shock with long-lasting ventilator care. Furthermore, recurrent duodenal ulcer bleeding might also have been a possible triggering factor for the disastrous manifestation in this patient.
The treatment for acute hyperammonemia due to OTCD encompasses not only decreasing intracranial pressure but also reducing the production of nitrogenous wastes and lowering the serum ammonia levels as quickly as possible. Concretely, this includes restricting dietary protein intake and facilitating the removal of ammonia via alternative pathwaysby the intake of oral sodium phenylbutyrate or sodium benzoate. Hemodialysis is also an effective means of lowering the serum ammonia level quickly, and recent guidelines recommend starting hemodialysis as early as possible, particularly if the serum ammonia level exceeds 200 umol/L despite an unconfirmed diagnosis of OTCD.[12] According to one case report, the MARS (extracorporeal liver support–albumin dialysis) was successful in lowering dialysis-unresponsive hyperammonemia in a patient with acquired hepatic glutamine synthetase deficiency after liver transplantation.[13] In this case, the patient also received continuous CRRT and MARS. However, the patient was not recovered, as severe cerebral edema and hypoxic brain damage had already developed. Other important treatments involve eliminating factors that precipitate hyperammonemia as promptly as possible, such as infection, GI bleeding, or medications (allopurinol, valproic acid). Associated factors in this patient were uncontrolled infection in addition to GI bleeding. Failure to remove these factors might have been another important cause of defeat. Liver transplantation is the last therapeutic option when the diagnosis of OTCD is definitely confirmed and life-threatening hyperammonemic crisis cannot be resolved despite ongoing treatments, although such a procedure may be accompanied by numerous complications.[11,14] Nonetheless, to prevent cognitive defects and developmental delay as a consequence of recurrent hyperammonemic episodes, liver transplantation is worth consideration as early as possible for children, whereas adult-onset OTCD does not have any widely accepted consensus regarding the indications or the best timing for liver transplantation.[9,14]
In summary, we have reported a very rare and unique case of life-threatening hyperammonemia caused by adult-onset OTCD, triggered by severe septic shock with acute respiratory failure in an immunocompromised host. This case suggests that if a patient presents with disproportionately high level of serum ammonia during ICU care and no apparent cause of hyperammonemia can be identified, intensivists should suspect an inborn error of metabolism such as UCD, and appropriate management should be started to lower the serum ammonia level as quickly as possible, even before the diagnosis is made.
NOTES
-
No potential conflict of interest relevant to this article was reported.
Fig. 1.Chest imaging (A, B) at admission shows bilateral patch consolidations, ground glass opacities and interlobular septal thickening with a small quantity of pleural effusion.
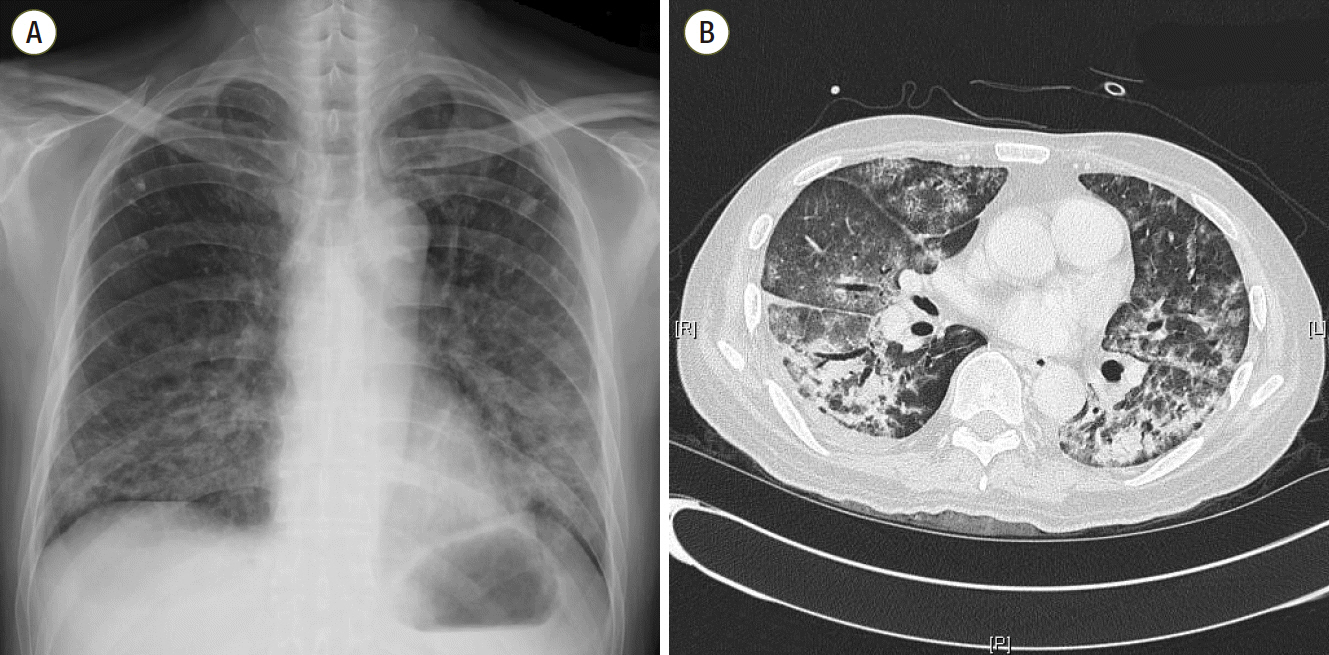
Fig. 2.Contrast brain magnetic resonance imaging shows severe brain swelling and increased cortical and subcortical gray matter signal changes suggesting hypoxic damage.
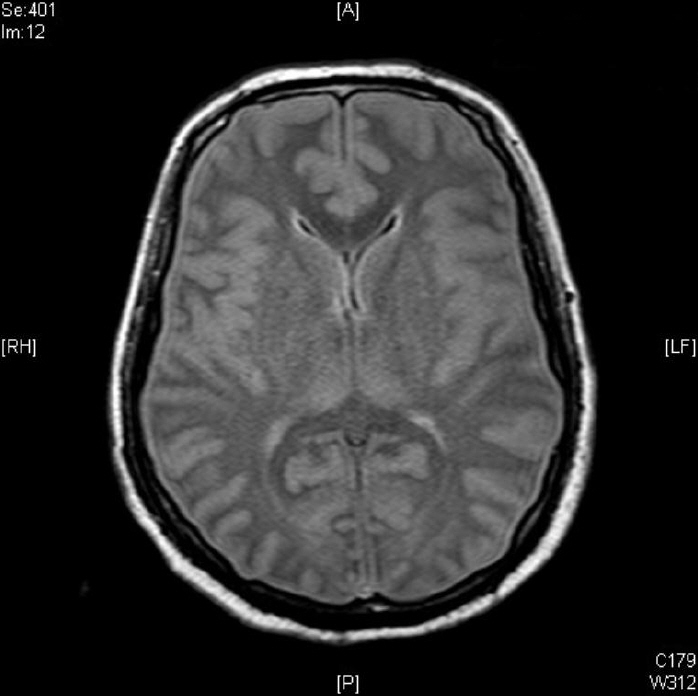
Fig. 3.Urine collection analysis displays urine orotic acid peak confirming the diagnosis of ornithine transcarbamylase deficiency (OTCD). OTCD: ornithine transcarbamylase deficiency.
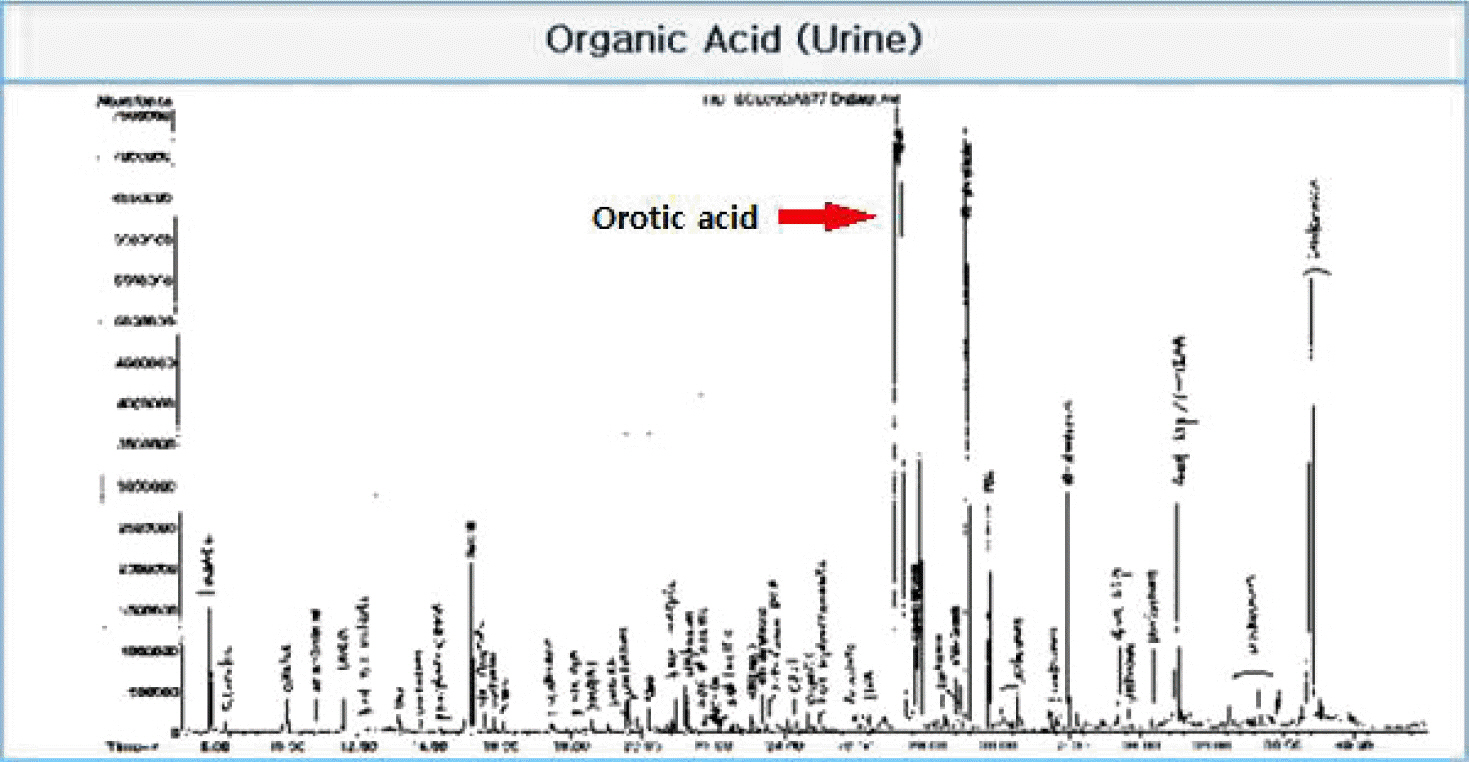
Table 1.Change in the levels of serum complete blood cell count, liver function test, serum BUN, creatinine, and ammonia
|
ER admission |
Hospital day 41 |
Hospital day 44 |
|
Hemoglobin (g/dL) |
11.2 |
8.5 |
8.8 |
|
White blood cell (/uL) |
1,550 |
5,350 |
8,380 |
|
Platelet(/uL) |
240,000 |
57,000 |
103,000 |
|
Bilirubin (mg/dL) |
0.3 |
0.3 |
0.2 |
|
AST (IU/L) |
22 |
45 |
19 |
|
ALT (IU/L) |
18 |
36 |
13 |
|
PT (sec)/INR |
12.2/1.03 |
14.1/1.19 |
16.6/1.39 |
|
BUN (mg/dL) |
14.9 |
71.6 |
72.2 |
|
Creatinine (mg/dL) |
1.56 |
1.88 |
3.02 |
|
Ammonia (µg/dL) |
|
411 |
1,700 |
References
- 1. Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis 2007;30:865-79.ArticlePubMedPMC
- 2. Shawcross D, Jalan R. The pathophysiologic basis of hepatic encephalopathy: central role for ammonia and inflammation. Cell Mol Life Sci 2005;62:2295-304.ArticlePubMedPMC
- 3. Vaquero J, Chung C, Cahill ME, Blei AT. Pathogenesis of hepatic encephalopathy in acute liver failure. Semin Liver Dis 2003;23:259-69.ArticlePubMed
- 4. Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest 2007;132:1368-78.ArticlePubMed
- 5. Bachmann C. Inherited hyperammonemia. In: Physicians Guide to the Laboratory Diagnosis of Metabolic Diseases. 2nd ed. In: Blau N, , Duran M, , Blaskovics ME, , Gibson KM. Berlin, Springer. 2003, pp 261-76.
- 6. Brusilow SW, Horwich AL. Urea cycle enzymes. In: The Metabolic and Molecular and Bases of Inherited Disease, 4 volume. 8th ed. In: Scriver CR, , Beaudet AL, , Valle D, , Sly WS, , Childs B, , Kinzler KW, , . New York, McGraw-Hill. 2000, pp 1909-61.
- 7. Yamaguchi S, Brailey LL, Morizono H, Bale AE, Tuchman M. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum Mutat 2006;27:626-32.ArticlePubMed
- 8. Arranz JA, Riudor E, Marco-Marín C, Rubio V. Estimation of the total number of disease-causing mutations in ornithine transcarbamylase (OTC) deficiency. Value of the OTC structure in predicting a mutation pathogenic potential. J Inherit Metab Dis 2007;30:217-26.ArticlePubMed
- 9. Wakiya T, Sanada Y, Mizuta K, Umehara M, Urahasi T, Egami S, et al. Living donor liver transplantation for ornithine transcarbamylase deficiency. Pediatr Transplant 2011;15:390-5.ArticlePubMed
- 10. Yorifuji T, Muroi J, Uematsu A, Tanaka K, Kiwaki K, Endo F, et al. X-inactivation pattern in the liver of a manifesting female with ornithine transcarbamylase (OTC) deficiency. Clin Genet 1998;54:349-53.ArticlePubMed
- 11. Gordon N. Ornithine transcarbamylase deficiency: a urea cycle defect. Eur J Paediatr Neurol 2003;7:115-21.ArticlePubMed
- 12. Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 2012;7:32. ArticlePubMedPMC
- 13. Chiu A, Tam S, Au WY, Chan SC, Liu CL, Fan ST. MARS treatment for a patient presenting with acquired hepatic glutamine synthetase deficiency after orthotopic liver transplantation. Liver Transpl 2005;11:353-5.ArticlePubMed
- 14. Busuttil AA, Goss JA, Seu P, Dulkanchainun TS, Yanni GS, McDiarmid SV, et al. The role of orthotopic liver transplantation in the treatment of ornithine transcarbamylase deficiency. Liver Transpl Surg 1998;4:350-4.ArticlePubMed
Citations
Citations to this article as recorded by





 PubReader
PubReader ePub Link
ePub Link Cite
Cite



