Dear Editor:
We read with immense interest the recent published narrative review by Chacón-Aponte et al. [1] which concluded that brain-lung interactions can affect patients with traumatic brain injury (TBI). This interaction leads to a cruel cycle that aggravates patient prognosis. According to these authors [1], the mechanisms of the interaction are complex and not fully explained. These authors [1] proposed two theories, the “blast injury” theory or “double hit” model, to represent the basis of its development and progression. The authors explained that the brain and lungs strongly interact through complex pathways from the brain to the lungs and vice versa.
For Chacón-Aponte et al. [1], the main pulmonary disorders that occur after brain injury are neurogenic pulmonary edema (NPE), acute respiratory distress syndrome, and ventilator-associated pneumonia, and the major brain consequences after lung injury can be explained by brain hypoxia and associated intracranial hypertension. Consequently, all of these conditions should be considered as management therapies after TBI and require special monitoring to avoid neurological and pulmonary complications. Although this review described brain-lung and lung-brain interactions with their pathophysiological and clinical consequences, we determined that two major consequences were not detailed or discussed in this narrative review, namely, the role of cardiac dysfunction and post-traumatic pulmonary embolism.
We agree with Chacón-Aponte et al. [1] that in patients with an acute traumatic head injury, altered pulmonary function is a frequent complication. NPE is a probable early contributor to the pulmonary dysfunction that occurs in patients suffering from acute head injuries. The diagnosis of NPE requires the exclusion of cardiogenic PE. The mechanism of NPE is not fully explained, and, to date, two theories have been developed to explain its occurrence: increased lung capillary permeability and increased pulmonary vascular hydrostatic pressure due to sympathetic overflow. It has been observed that catecholamine storms lead to a state of systemic vasoconstriction with strong venoconstriction, resulting in the induction of elevated pulmonary artery wedge pressure and hydrostatic PE. However, besides these two hypotheses, NPE can also result from cardiac dysfunction [2]. Therefore, we propose a new hypothesis that cardiac dysfunction is due to TBI rather than PE due to existing chronic heart failure.
In one local study involving seven patients (with no history of cardiorespiratory disease) with NPE following head trauma, we found that all the patients developed a clinical, echocardiographic, and/or histological signs of cardiac dysfunction. In fact, evidence of myocardial dysfunction was confirmed on echocardiography in three patients and pulmonary artery catheterizations in three patients. Moreover, cardiac damage was confirmed by post-mortem myocardial biopsies in four patients. Echocardiography studies, repeated for two patients within 7 and 90 days after trauma, exhibited complete improvement in wall motion with a full recovery of left ventricular ejection fractions in these patients.
Moreover, in a retrospective study including 20 patients with NPE requiring mechanical ventilation, Deehan and Grant [3] determined that cardiac index and left ventricular stroke work index (LVSWI) were markedly depressed with an increase of pulmonary artery wedge pressure (17 mm Hg) in 12 of the 20 patients. In the same study [3], mean systemic vascular resistance index was significantly increased. The use of dobutamine was associated with a significant increase in the cardiac index and LVSWI. These authors [3] concluded that NPE was generally associated with myocardial dysfunction reversed through the use of dobutamine. This myocardial dysfunction can be related to catecholamine storm, hyperglycemia, and/or the massive liberation of cytokines usually observed after severe TBI [2]. These phenomena can be complicated by cardiac ischemia, arrhythmia, and negative inotropic effects on the heart, leading to heart failure and NPE [2]. This cardiac dysfunction with associated PE results in a decrease in organ blood flow and oxygen delivery to the brain. All these observable facts lead brain ischemia and can seriously affect patients with TBI.
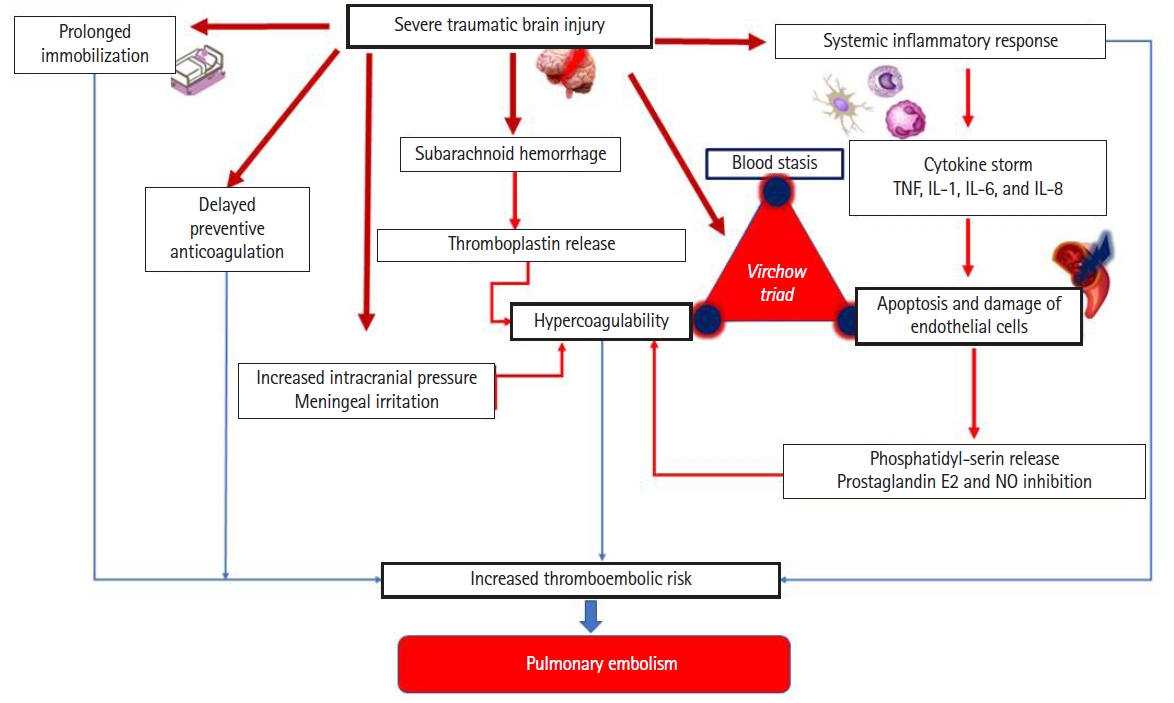
On the other hand, it is well documented that post-traumatic PE is a frequent complication of head trauma with an overall incidence ranging from 8% to 24% [4,5]. In particular, severe TBI significantly increases the risk of thromboembolic events, and PE in particular. The development of PE is associated with a poor outcome involving a high mortality rate and a long duration of stay in the intensive care unit and hospital [5]. Interestingly, recent literature suggests that patients are at risk for hypercoagulability early after the traumatic injury, leading to an early occurrence of PE (within 48 hours after trauma)[4]. In fact, on the physiopathological level, there are several factors explaining the development of PE after TBI. In addition to the risk factors for hypercoagulability described by Virchow (stasis due to prolonged immobilization, hypercoagulability, and endothelial injury), the role of inflammation through endothelial damage in this specific condition should be considered. In fact, although trauma-patients diverge according to the type of injury (location and the presence of cranial injuries), severe TBI are associated with an increase at the level of tissue factors, pro-inflammatory procoagulant cytokines (tumor necrosis factor, interleukins [IL]-1, IL-6, and IL-8), and markers of thrombin generation [4,5]. Moreover, in severe TBI, the levels of physiological anticoagulants (antithrombin, protein C, and protein S) are reduced leading to the development of venous thrombo-embolic complications. Figure 1 explains the mechanisms of the development of PE in patients suffering from TBI. Moreover, based on the increased risk of aggravation of intracranial bleeding in TBI patients, the initiation of pharmacological prophylaxis is commonly delayed. Therefore, delayed initiation may increase the risk of PE [4,5]. Acute PE increases pulmonary vascular resistance and right ventricular afterload. The immediate consequences are hypoxemia, pulmonary vasoconstrictors, and/or cardiogenic shock, leading to brain ischemia and/or hypoxic damage. As a consequence, the development of PE for patients suffering from TBIs is associated with a poor outcome of a high mortality rate, a high risk of nosocomial infections, and a greater prolonged length of stay in the intensive care unit and hospital [5].
In conclusion, we congratulate Chacón-Aponte et al. [1] on their narrative review, which concluded that the brain-lung interaction can seriously affect patients with TBI, initiating a vicious cycle that aggravates patient prognosis. However, we found that the role of cardiac dysfunction due to TBIs and post-traumatic pulmonary embolisms should be included with the other mechanisms detailed by these investigators.
NOTES
-
CONFLICT OF INTEREST No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conceptualization: MB, MB. Data curation: all authors. Writing–original draft: MB (Mabrouk Bahloul). Writing–review & editing: all authors.
Figure 1.Mechanisms of development of pulmonary edema in patients suffering from traumatic brain injury. TNF: tumor necrosis factor; IL: interleukin; NO: nitric oxide.

References
- 1. Chacón-Aponte AA, Durán-Vargas ÉA, Arévalo-Carrillo JA, Lozada-Martínez ID, Bolaño-Romero MP, Moscote-Salazar LR, et al. Brain-lung interaction: a vicious cycle in traumatic brain injury. Acute Crit Care 2022;37:35-44.ArticlePubMedPMC
- 2. Bahloul M, Chaari AN, Kallel H, Khabir A, Ayadi A, Charfeddine H, et al. Neurogenic pulmonary edema due to traumatic brain injury: evidence of cardiac dysfunction. Am J Crit Care 2006;15:462-70.ArticlePubMed
- 3. Deehan SC, Grant IS. Haemodynamic changes in neurogenic pulmonary oedema: effect of dobutamine. Intensive Care Med 1996;22:672-6.ArticlePubMed
- 4. Bahloul M, Dlela M, Bouchaala K, Kallel H, Ben Hamida C, Chelly H, et al. Post-traumatic pulmonary embolism: incidence, physiopathology, risk factors of early occurrence, and impact outcome: a narrative review. Am J Cardiovasc Dis 2020;10:432-43.PubMedPMC
- 5. Bahloul M, Chelly H, Regaieg K, Rekik N, Bellil S, Chaari A, et al. Pulmonary embolism following severe traumatic brain injury: incidence, risk factors and impact outcome. Intensive Care Med 2017;43:1433-35.ArticlePubMed
Citations
Citations to this article as recorded by


 , Karama Bouchaala, Najeh Baccouche, Kamilia Chtara, Hedi Chelly, Mounir Bouaziz
, Karama Bouchaala, Najeh Baccouche, Kamilia Chtara, Hedi Chelly, Mounir Bouaziz 

 PubReader
PubReader ePub Link
ePub Link Cite
Cite