Sepsis-induced cardiac dysfunction: a review of pathophysiology
Article information

Abstract
It is well known that cardiac dysfunction in sepsis is associated with significantly increased mortality. The pathophysiology of sepsis-induced cardiac dysfunction can be summarized as involving impaired myocardial circulation, direct myocardial depression, and mitochondrial dysfunction. Impaired blood flow to the myocardium is associated with microvascular dysfunction, impaired endothelium, and ventriculo-arterial uncoupling. The mechanisms behind direct myocardial depression consist of downregulation of β-adrenoceptors and several myocardial suppressants (such as cytokine and nitric oxide). Recent research has highlighted that mitochondrial dysfunction, which results in energy depletion, is a major factor in sepsis-induced cardiac dysfunction. Therefore, the authors summarize the pathophysiological process of cardiac dysfunction in sepsis based on the results of recent studies.
INTRODUCTION
Sepsis-induced cardiac dysfunction refers to a wide spectrum of acute myocardial impairment caused by sepsis [1]. Widely understood as a systemic inflammatory response syndrome that occurs during infection, sepsis takes place due to a dysregulated immune response to infectious agents [2]. Myocardial dysfunction in sepsis has become a focus of many studies during the last five decades (Figure 1). The presence of myocardial dysfunction in sepsis is known to be associated with significantly increased mortality of 20% to 50% [3].
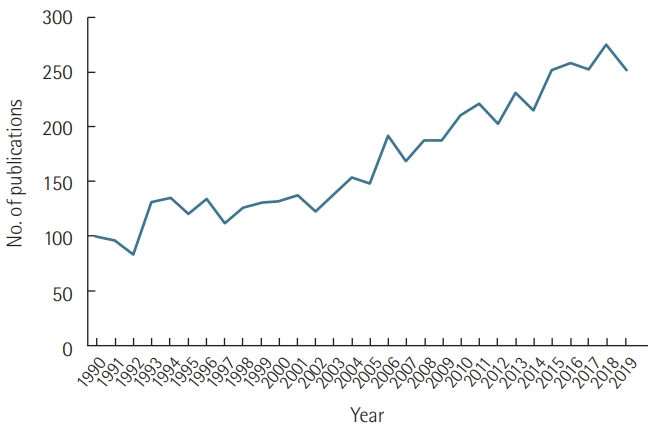
The number of publications per year on sepsis-induced cardiac dysfunction, obtained by searching for “sepsis,” “cardiac dysfunction,” “myocardial dysfunction,” and “cardiomyopathy” in PubMed. The number was calculated from January 1, 1990 to December 31, 2019.
Despite significant efforts made in studying its clinical presentation and in exploring its pathophysiological mechanisms and diagnostic options, septic cardiomyopathy remains a serious medical conundrum of which researchers’ understanding remains insufficient, with no definitive therapeutic option available to date [4]. Several recent studies have addressed the issue of uncertainty in classifying alterations of impaired myocardial function in sepsis and in describing its underlying mechanisms. However, there is well-established evidence associating sepsis-induced impairment of cardiovascular function with a weakened response of the host’s immune system to the invading microorganisms. This results in an inflammatory reaction, which is intensified by inflammatory mediators released into the bloodstream and provokes hemodynamic changes that lead to impaired cardiac function [5].
Even though several biochemical mediators and pathways have received considerable attention in the literature, the specific cause of myocardial depression in sepsis remains unclear. Understanding the pathophysiology of sepsis-induced cardiac dysfunction has been a challenging task for both clinicians and researchers. Therefore, this review provides an essential understanding of myocardial dysfunction in sepsis by summarizing current knowledge regarding the pathophysiological processes behind sepsis-induced cardiac dysfunction.
CLINICAL MANIFESTATIONS OF SEPSISINDUCED CARDIAC DYSFUNCTION
Contractile dysfunction is the main characteristic of cardiac dysfunction in sepsis, and it manifests as biventricular dilatation, a reversible decrease in ejection fraction, a diminished blood pressure response to intravenous fluid resuscitation, and an inability to improve cardiac output despite high levels of circulating catecholamines [1]. The clinical presentation of myocardial dysfunction can involve one or all of the following: left ventricular (LV) systolic dysfunction, LV diastolic dysfunction, right ventricular (RV) impairment, and/or dilatation of both ventricles [6]. Different studies have presented pump failure as result of dramatic afterload reduction associated with lowered systemic vascular resistance (SVR) and decreased cardiac output/cardiac index, a condition that causes regional and even global contractile impairment [7]. LV and RV dilatation resulting from an increase of compliance is always considered as a sign of improvement. Meanwhile, RV dysfunction is known to have an effect on the LV. RV dilata- tion reduces the ventricular ejection fraction and impairs LV performance by decreasing the LV filling pressure and by causing mechanical compromise due to the shift of the LV septum.
Cardiac dysfunction was once considered to occur only during the hypodynamic phase of shock characterized by increased SVR, tissue hypoperfusion, cold skin, and eventual organ failure, but it was later proven that myocardial impairment also occurs during hyperdynamic septic shock, which manifests as increased cardiac output, decreased SVR, and warm, perfused skin [8]. The LV dilatation in hyperdynamic septic shock is considered to be an adaptive response that preserves cardiac output via the Starling mechanism, which was found to be associated with lower mortality and improved prognosis in patients with sepsis [9]. The pathophysiology involved in LV systolic dysfunction in septic patients was confirmed to range from isolated diastolic dysfunction to combined systolic and diastolic abnormalities [10]. Septic patients were found to have a reduced LV ejection fraction with acute LV dilatation manifested by an increased LV end-diastolic volume index in comparison with controls [11]. RV systolic dysfunction involves a decreased RV ejection fraction and RV dilatation. Based on the fact that RV dysfunction occurs independently of changes in peripheral vascular resistance and pulmonary arterial pressure, some studies have suggested that an increased RV afterload could not be the main cause of RV depression in septic shock. Even though sepsis-associated RV dilatation is known to cause septal displacement, thereby decreasing LV compliance, preload, and general performance, it has also been stressed that this finding is not common [12]. Despite the differences between the ventricles in structure and function, recent evidence suggests that sepsis-induced RV dysfunction is broadly similar to LV dysfunction.
PATHOPHYSIOLOGY OF SEPSIS-INDUCED CARDIAC DYSFUNCTION
Our knowledge of sepsis-induced cardiac dysfunction depends on our understanding of sepsis itself. The pathophysiology of sepsis consists of an uncontrolled inflammatory reaction resulting from defective interactions between the immune system and invading pathogens. In a normal individual, the host immune system usually identifies a pathogenic invasion by the use of pattern recognition receptors (PRRs), which bind to pathogen-associated molecular patterns (PAMPs). PRRs, which include Toll-like receptors (TLRs), are expressed on the surface of the immune cells of the host, whereas PAMPs, such as lipopolysaccharide (LPS), lipoteichoic acid, and others, belong to the invading microorganisms. In addition to their role of initiating the host immune system defense against pathogenic invasions and in mediating the recognition of PAMPs, PRRs are also able to identify and bind to danger-associated molecular patterns (DAMPs), which are released during the inflammatory reaction, to activate the innate immune system [13].
Although it has been the focus of many studies for decades, our understanding of the pathophysiology of septic cardiomyopathy remains incomplete. In recent years, cytokines have been identified as the main mediators of myocardial depression in sepsis due to their release, which is triggered by the body’s response to surgical, traumatic, ischemic, or septic insults [14]. However, septic cardiomyopathy can also be induced by many other factors, making it necessary to explore individual contributing mechanisms in order to generate appropriate therapeutic targets (Figure 2).
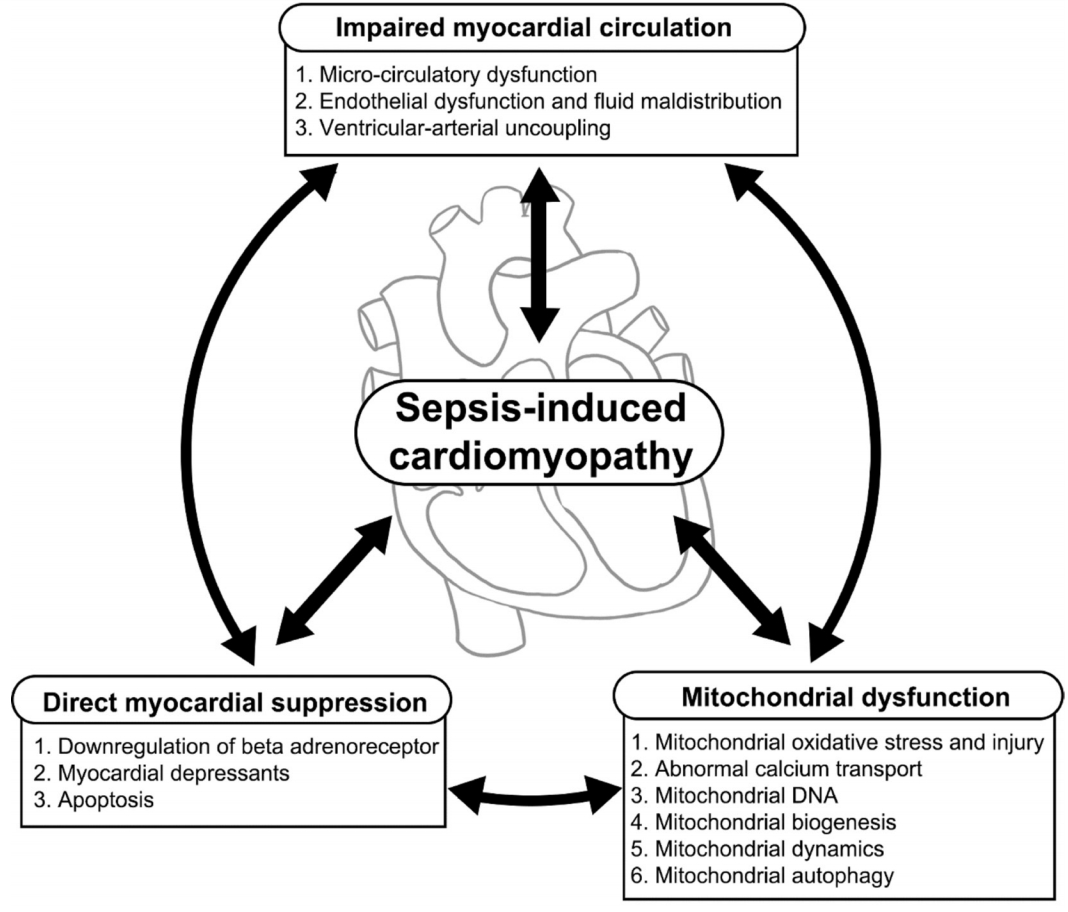
Pathophysiology of sepsis-induced cardiac dysfunction.
Myocardial Ischemia
Reduced blood flow to the myocardium is among the most important mechanisms of sepsis-induced cardiac dysfunction. The reversible nature of myocardial depression was found to be related to the concept of myocardial hibernation in sepsis, which is an adaptive response to ischemia and hypoxia, and was proposed as common manifestation of myocardial dysfunction in sepsis [15]. Despite this protective mechanism, ischemia of the cardiac muscle can severely aggravate septic myocardial dysfunction in patients with severe sepsis. Cardiac function is strongly dependent on intravascular fluid status. The loss of vascular tone secondary to arterial vasodilation is among the main factors behind hemodynamic instability in septic patients. Newly identified physiological factors include cardiac contractility, end-systolic ventricular elastance (Ees), arterial elastance (Ea), LV systolic performance, as well as ventriculo-arterial coupling, defined as the ratio of Ea to Ees, which is an important element in evaluating the efficiency of the cardiovascular system.
While the endothelium plays a prominent role in sepsis, little is known regarding the impact of sepsis-associated endothelial dysfunction. In a previous study, even though microcirculatory coronary blood flow was maintained at an adequate level or even higher in patients with septic shock, optimal coronary blood flow did not prevent myocardial dysfunction due to septic endothelial damage, which caused blood flow maldistribution; this finding provides strong support for the idea that there are multiple factors behind septic cardiomyopathy [16]. Although the above findings reflect important alterations in coronary flow and myocardial metabolism, the evidence that myocardial ischemia alone is the underlying cause of myocardial dysfunction in sepsis is still weak.
Direct Myocardial Suppression
The first biochemical mechanism behind direct myocardial depression consists of a decrease of β-adrenergic receptor components, which impairs the adrenergic response at the cardiomyocyte level in a process mediated by various pro-inflammatory substances (mainly cytokines and nitric oxide [NO]). The second mechanism is characterized by cardiomyocyte injury and/or death caused by toxins, complements, DAMPs, and other myocardial depressants [17]. Thirdly, apoptosis of cardiomyocytes is among the leading causes of myocardial depression and sepsis-induced multiple organ dysfunction. When not treated early, myocardial dysfunction often progresses with apoptosis of cardiomyocytes, followed by downregulation of β-adrenoceptors and impairment of myofibril function due to the disruption of calcium liberation.
Cytokines
Cytokines are peptides occasionally produced locally from both somatic cells (endothelial, epithelial, fibroblasts) and immune cells (neutrophils, lymphocytes, macrophages) in response to stress or challenges due to surgical insults, trauma, ischemia, or sepsis, and their signal serves as a means of communication among challenged cells [18]. Cytokines are also known to induce the release of additional inflammatory factors such as prostanoids, NO, and many others, which eventually contribute to myocardial dysfunction. Tumor necrosis factor (TNF)-α and interleukin (IL)-1β are the most common pro-inflammatory cytokines. They are released by macrophages in sepsis and have demonstrated significant cardiac contractility depression activity in vitro [19]. While TNF-α and IL-1β are considered to be the most important components of the inflammatory mechanism [20], NO and oxygen-free radicals are regarded as second-degree factors in the process of septic myocardial depression [21]. Synthesized in response to TNF-α, IL-1 depresses cardiac contractility by stimulating NO synthase (NOS) [22]. Therefore, inhibitors of IL-1 such as IL-1 receptor antagonist could be a good choice to reduce the morbidity of septic cardiomyopathy patients and to improve their survival, but this finding lacks enough supporting evidence [23].
An increased cytokine level induces the release of more cytokines and other chemical mediators, making the process of myocardial dysfunction more complex and severe. The heart was also shown to be a cytokine producer during inflammation, thereby intensifying myocardial depression and cardiac function impairment. This was observed when IL-6 was produced in the heart due to the stimulation of myocardial α- and β-adrenoreceptors and from the excess use of catecholamines [24]. Increased circulating serum levels of IL-6 were found to be associated with the severity of illness and the degree of vasopressor requirement in septic shock patients [25].
Nitric oxide
Expressed in the vascular endothelium, NO is a potential mediator of septic cardiomyopathy and has a wide range of physiological effects in the cardiovascular system [26]. NO is produced by various isoforms of NOS that exist in subcellular components, and it acts as a second messenger within these components. NOS 1 and 3 were shown as potential players in early septic cardiomyopathy, while NOS 2 was identified as a possible cause of contractile depression in late sepsis [27]. Several studies have found NO to be associated with the severity of cardiac dysfunction and a higher mortality rate due to its effects on various sites, including the β-adrenergic receptors of the heart, as well as at the mitochondrial level, where it induces mitochondrial function impairment, one of the major pathways of sepsis-induced cardiomyopathy [28].
Prostanoids
Elevated levels of thromboxane, prostacyclin, and other prostanoids have been found in the serum of septic patients. Prostanoids have been proven to have the potential to alter coronary endothelial function, but their effect can be diminished by cyclooxygenase inhibitors such as indomethacin. Prostanoid inhibition by nonsteroid anti-inflammatory drugs, mainly ibuprofen and lornoxicam, has also been proposed as a treatment option, but neither drug was efficient in improving survival in clinical studies [29].
Endothelin-1
High levels of endothelin-1 (ET-1) were found to be able to trigger the release of inflammatory cytokines. Endothelin-receptor blockade with tezosentan improved cardiac performance in a porcine endotoxic model [30]. The role of ET-1 in septic myocardial dysfunction is not widely supported. Therefore, further exploration is necessary to assess its mechanism of action.
Intracellular adhesion molecules
The increased expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) was observed in coronary endothelium and in cardiomyocytes after LPS and TNF-α stimulation in mice [31]. The blockade of VCAM-1 prevented myocardial dysfunction and decreased myocardial neutrophil accumulation [32]. Furthermore, both knockout and antibody blockade of ICAM-1 ameliorated myocardial dysfunction in endotoxemia without affecting neutrophil accumulation. Even though the antibody blockade of either ICAM-1 or VCAM-1 abolished contractile dysfunction, further research is suggested to understand the effect of adhesion molecules on calcium homeostasis and the generation of oxygen free radicals.
Complement system
The humoral immune response becomes activated in sepsis, triggering a cascade of complement proteins such as C5a, a powerful pro-inflammatory agent known to increase neutrophil chemotaxis, granular enzyme release, reactive oxygen species (ROS) generation, and synthesis of cytokines. Additionally, the expression of C5aR in cardiomyocytes mediates C5a-induced cardiodepression, which can be reversed by administration of anti-C5a antibody [33].
Histones and high mobility group box 1 (HMGB1)
High levels of histones were found to be associated with newonset LV dysfunction and a higher prevalence of new-onset arrhythmias in a mouse septic model and in human sepsis [34]. Circulating histones were also correlated with sepsis severity and outcome. HMGB1 is a DAMP released during tissue damage that acts by amplifying oxidative stress through HMGB1–TLR4 interactions and by impairing cardiac excitation- contraction coupling [35]. DAMPs have a direct damaging effect on cardiomyocytes in a vicious cycle wherein injured myocytes produce more DAMPs, which intensify myocardial dysfunction in sepsis.
Other mediators
The evidence for more myocardial depressant substances is still emerging, with many new endogenous substances being identified as potential causes of septic myocardial depression. These include estrogenic compounds, histamine, eicosanoids/ prostaglandins, and leukocyte lysozyme [36]. However, some of these newly discovered substances, such as endotoxin and natriuretic peptides, still need further exploration. Both B-type natriuretic peptide and atrial natriuretic peptide were significantly elevated in septic patients when compared to controls [37]. Caspase-3 activation was reported to be associated with decreased sensitivity of the myofilaments to calcium, as well as structural breakdown of the sarcomeres, but further studies are necessary to clarify this. Additional data have also suggested that platelet-activating factor, cyclooxygenase products, leukotrienes, protein kinase C, and lipoteichoic acid may be involved in septic myocardial depression [38].
Metabolic Dysfunction in Mitochondria
Even though the mechanisms associated with mitochondrial dysfunction in sepsis are not fully understood, several studies have confirmed that mitochondrial dysfunction is a key player in septic cardiomyopathy through energy depletion [39,40]. The fact that the heart is a mitochondrial-rich organ explains why mitochondrial dysfunction is strongly associated with sepsis-induced cardiac dysfunction and the prognosis thereof [41]. The heart is likely to be affected by sepsis-induced mitochondrial dysfunction due to its strong dependence on high adenosine triphosphate (ATP) levels to maintain its contraction and diastolic function. With the aim of finding options of restoring mitochondrial function, various mechanisms have been explored, especially mitochondrial uncoupling proteins located in the mitochondrial inner membrane, which promote proton leakage across the mitochondrial inner membrane, an essential regulator of mitochondrial membrane potential and a generator of ROS and ATP. Other involved biochemical pathways include oxidative stress, mitochondria Ca2+ flux, mitochondrial DNA (mtDNA) in sepsis, mitochondrial dynamics, mitochondrial biogenesis, and mitochondrial autophagy.
Mitochondrial oxidative stress and injury
Levels of ROS such as superoxide are increased due to mitochondrial dysfunction in sepsis, leading to oxidative injury and causing continuous damage to cells and organs [42]. An unbalanced production of ROS and reactive nitrogen species impairs oxidative phosphorylation in the septic myocardium [43]. This directly inhibits mitochondrial respiration and damages several sub-cellular components, such as mitochondrial proteins, often leading to mitochondrial apoptosis. Oxidative stress results in mitochondrial dysfunction and has been strongly associated with organ failure in sepsis models and reported in septic patients [44]. Due to oxidative stress, the antioxidant capacity is diluted because of decreased levels of antioxidants like vitamin C and E, unconjugated bilirubin, uric acid, and others [45]. It is therefore clear that oxidative stress plays an indisputable role in the pathophysiology of septic myocardial dysfunction, which suggests that antioxidants may be a potential therapeutic option in the future [46,47].
Abnormal calcium transport in mitochondria
One of the most important steps in regenerating ATP is building up a proton gradient, which is dependent on the impermeability of the inner mitochondrial membrane; in turn, this is mainly mediated by Ca2+ [48,49]. The release of chemical mediators such as cytokines during sepsis raises the mitochondrial Ca2+ content by increasing Ca2+ leakage from the sarcoplasmic reticulum and decreasing Ca2+ uptake into the same organelle [50]. Ca2+ overload causes mitochondrial permeability transition pores (mPTPs) to open and triggers sequential pathological changes, such as the activation of caspase proteins, causing irreversible damage to the mitochondria that eventually leads to cardiomyocyte contractile dysfunction [51,52]. When cytoplasmic Ca2+ levels diminish, smooth muscle relaxation and consequent vasodilation occur. When this process happens in cardiac cells, it results in decreased myocyte contraction and, therefore, impaired myocardial function [53]. Based on the crucial role of Ca2+ in myocardial contraction, various studies have explored its involvement in septic myocardial depression and concluded that several pathways such as impaired Ca2+ uptake, impaired Ca2+ release from sarcoplasmic reticulum storage, and decreased Ca2+ channel sensitivity are all involved in sepsis-related cardiac depression [54]. Abnormal transport of Ca2+ is therefore an important factor affecting heart function.
Mitochondrial DNA
Based on its ability to act as a DAMP, mtDNA, which is a circular molecule that encodes the key proteins involved in the oxidative phosphorylation system, has become the focus of considerable research [55,56]. In addition to its coding function, mtDNA is also involved in cellular immune functions [57]. Multiple in vivo and in vitro studies have shown that mtDNA can be transferred from mitochondria to the cytosol via mPTPs, and for this reason, any pathological changes causing mPTP opening would increase the leakage of mtDNA [58]. In a study of mtDNA in intensive care unit patients, it was found that the levels of circulating mtDNA were significantly higher in nonsurvivors than in survivors [59]. Another study also found that plasma mtDNA levels in patients with sepsis were higher than in healthy controls [60]. From this evidence, it was later demonstrated that a high concentration of mtDNA led to increased neutrophil viability, which was proven to be associated with poor outcomes in patients with sepsis.
Mitochondrial dynamics
Mitochondria frequently undergo fission and fusion events to modulate mitochondrial morphology, number, and size. In physiological conditions, mitochondrial fusion and fission is balanced to maintain mitochondrial and cellular homeostasis by regulating cell division and proliferation, as well as in the self-directed removal of damaged mitochondria. Mitochondrial membrane fusion and fission depend on proteins encoded by different genes, such as mitofusin-1 (encoded by MFN1) and mitofusin-2 (MFN2) in membrane fusion, and mitochondrial dynamin-like GTPase (encoded by OPA1) linked with membrane fission [61]. These proteins have been associated with altered mitochondrial membrane potential and reduced oxygen consumption. Fission and fusion increase in stressful conditions, playing critical roles in removing damaged mitochondria and augmenting repair processes [62]. Even though these proteins appear to play a critical role in fission and fusion processes, their exact functions remain unclear. During sepsis, mitochondrial dysfunction results in activation of mitochondrial fission and inactivation of mitochondrial fusion, which promotes dysfunctional mitochondrial fragmentation and ultimately results in organ failure [63]. Using pharmacologic therapeutic agents to activate mitochondrial biogenesis or mitochondrial autophagy, or to decrease mitochondrial fission, in a septic model was also proven to ameliorate organ mitochondrial dysfunction and organ failure [64].
Mitochondrial biogenesis
Mitochondrial biogenesis involves the production of mitochondrial proteins encoded either by nuclear DNA, with subsequent import and integration into the mitochondria, or via mtDNA, which encodes proteins mainly situated within the oxidative phosphorylation pathway. Biogenesis thus replaces damaged proteins and improves the capacity for energy production if energy demands increase over time [65]. Therefore, mitochondrial biogenesis is an important mechanism for meeting the energy needs of cell metabolism.
Mitochondrial autophagy
Removal of dysfunctional mitochondria is another key mechanism of organ recovery from sepsis. Autophagy is a central process in cell survival under stress, as it eliminates damaged proteins and organelles by formation of a double membrane autophagosome, which then fuses with lysosomes. Mitophagy, which refers to the selective autophagy of mitochondria, requires receptors to recognize damaged mitochondria for degradation. In sepsis, mitochondrial dysfunction induces a loss of mitochondrial membrane potential that triggers mitophagy. Even though mitochondrial autophagy is considered beneficial, impaired autophagy is also known to contribute to mitochondrial dysfunction and can lead to adverse outcomes during sepsis due to depletion of the mitochondrial population [66,67]. Mitophagy is currently being considered in many studies as a therapeutic target to improve heart function.
CONCLUSIONS
We reviewed the pathophysiology of sepsis-induced cardiac dysfunction but reaffirmed that its pathophysiology has not been fully elucidated. Despite being the object of intense research for many years, the treatment of sepsis-induced cardiac dysfunction remains a dilemma. Various clinical and experimental studies have been conducted on the treatment of sepsis-induced cardiac dysfunction, as well as sepsis more broadly, but many studies have reported ineffective or weak results in the verification phase. Therefore, we believe that a more accurate understanding of the pathophysiology of sepsis- induced cardiac dysfunction is an essential gateway to the development of more effective treatment methods.
KEY MESSAGES
▪ The major pathophysiology of sepsis-induced cardiac dysfunction can be summarized as follows: impaired myocardial circulation, direct myocardial depression, and mitochondrial dysfunction.
▪ Impaired blood flow to the myocardium is associated with microvascular dysfunction, impaired endothelium, and ventriculo-arterial uncoupling.
▪ Recent research highlights that mitochondrial dysfunction is a major factor in sepsis-induced cardiac dysfunction.
Notes
CONFLICT OF INTEREST No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conceptualization & Data curation: all authors. Visualization: DK, KL, IJ. Writing–original draft: RH, IC. Writing–review&editing: HJC, IJ.