The Anti-Inflammatory Effect of Arginine-Vasopressin on Lipopolysaccharide-Induced IκBα/Nuclear Factor-κB Cascade
Article information
Abstract
Background
Arginine vasopressin (AVP) is widely used as a vasopressor agent. Some recent studies have suggested that AVP may exert an immunomodulatory effect. However, the mechanism about the anti-inflammatory effect of AVP is not well known. We investigated the effect of AVP on the ihibitor of kappa B (IκBα)/nuclear factor-kappa B (NF-κB) pathway in RAW 264.7 cells.
Methods
Cultured RAW 264.7 cells were pretreated with AVP and stimulated with lipopolysaccharide (LPS). To evaluate the effect of AVP on inflammatory cytokines, the concentration of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) were assessed by an enzyme-linked immunosorbent assay technique. The expression of IκBα and nuclear translocation of NF-κB p65 were measured by Western blotting, and IκB kinase (IKK) activity was analyzed by an in vitro immune complex kinase assay. To confirm the AVP effect on IκBα/NF-κB cascade and via V2 receptor, we added tolvaptan (V2 receptor antagonist) after AVP pretreatment.
Results
The increase of IL-6 and TNF-α in LPS-stimulated RAW 264.7 cells was suppressed by a treatment with AVP. Pretreatment of AVP inhibited increasing of IKK activity and IκBα degradation induced by LPS in RAW 264.7 cells. Furthermore, LPS induced and NF-κB transcription was inhibited by AVP pretreatment. The observed changes in IKK activity, IκBα degradation and NF-κB transcription by AVP was abolished by tolvaptan treatment.
Conclusions
Our results suggest that AVP showed anti-inflammatory effect on LPS-induced IκBα/NF-κB cascade in mouse macrophages via V2 receptors.
Introduction
Sepsis is a life-threatening disease caused by excessive host immune response as a result of sustained infection. It can result in a severe systemic inflammatory response and, possibly, shock.[1,2] Despite significant improvements in critical care medicine recently, severe sepsis and septic shock remain as leading causes of morbidity and mortality among intensive care unit (ICU) patients.[2-4]
It is well known that nuclear factor-kappaB (NF-κB), a family of DNA-binding protein factors that are required for transcription of most proinflammatory molecules, has a potential role in systemic inflammatory response syndrome (SIRS).[5,6] Moreover, there has been several studies regarding some drugs that may have an anti-inflammatory effect through the NF-κB pathway.
Vasopressin is an emerging rational therapy for septic shock patients who exhibit a deficiency of endogenous vasopressin combined with an exquisite cardiovascular sensitivity to exogenous low-dose vasopressin.[7-10] It has been suggested that early use of vasopressors may exert an immunomodulatory effect in sepsis, in addition to the vasoactive effect.[11-13]
However, evidence in scarce in terms of analyzing the mechanism about immunomodulatory effect of vasopressin. Therefore, this experimental study was performed to investigate the effects of vasopressin on the activaion of the ihibitor of kappa B (IκBα)/NF-κB in mouse macrophage.
Materials and Methods
1) Cell culture
The murine macrophage/monocyte cell line RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC). These cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) 1640 medium (GIBCO BRL, Gaithersburg. MD, USA), containing 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin, at 37.8℃ under 5% CO2.
2) Reagents
[Arg8]-vasopressin solution, antibody to vasopressin V2 receptor (V2R), tolvaptan, a potent active non-peptide vasopressin V2 selective antagonist, and lipopolysaccharide (LPS) were purchased from Sigma Aldrich (St. Louis, MO, USA). NF-κB p65 was obtained from cell signaling technology (Danvers, MA, USA). Rabbit polyclonal anti-IκBα, goat polyclonal anti-actin, donkey anti-goat, and donkey anti-rabbit secondary antibody conjugated with horseradish peroxidase (HRP) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The enhanced chemiluminescence (ECL) kit was from Thermo Fisher Scientific Inc.
3) Preparation of cell extraction
The cytoplasmic and nuclear extracts were prepared using a commercially available Nuclear Extraction Kit (Active Motif) in accordance to the manufacturer’s instructions. In brief, we aspirated the media out of the cell dish and washed it with 5 mL ice-cold PBS/phosphatase inhibitor. Then, the aspirated solution was taken out and added 3 mL ice-cold PBS/phosphatase inhibitors. The cells were removed from the dish by gently scraping with cell lifter and transferred to a pre-chilled 15 mL conical tube. Then, centrifugation of cell suspension was performed for 5 minutes at 500 rpm in a centrifuge pre-cooled at 4℃, and we discarded the supernatant. We gently resuspended the cells in 500 uL 1 X Hypotonic Buffer by pipetting up and down for several times. Then, we transferred the cells to a pre-chilled microcentrifuge tube. After that, microcentrifuge tube was incubated for 15 minutes on ice. We then added 25 μL Detergent and vortexed it for 10 seconds at highest setting. We used a centrifuge for 30 seconds at 14,000 xg to separate the supernatant, which was transferred into a pre-chilled microcentrifuge tube and stored at -80℃ until ready to use. Then, a nuclear pellet was resuspended in 50 uL Complete Lysis Buffer by pipetting up and down, and we used a vortex for 10 seconds at highest setting. Subsequently, we incubated the suspension for 30 minutes on ice on a rocking platform set at 150 rpm, and then repeated the vortex for additional 30 seconds. Thereafter, we used a centrifuge for 10 minutes at 14,000 xg, transferred the supernatant into a pre-chilled microcentrifuge tube, and stored at -80℃. At last, protein concentrations were determined using the Bradford method (Bio-Rad).
4) Enzyme-linked immunosorbent assay (ELISA)
The cells (3×105) were grown on 6-well culture plates in equal numbers. The supernatants were collected and centrifuged for 5 minutes in a tabletop centrifuge to remove non-adherent cells and were immediately frozen at -70°C until analysis. The samples were subsequently analyzed for secreted cytokine concentrations, which were quantitated using an ELISA.
5) Western blot analysis
A 30 or 50 μg of protein was resolved by 10% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with 5% skim milk-pbs-0.1% Tween 20 for 1 hour at room temperature before being incubated overnight with primary antibodies diluted 1: 1,000 in 5% skim milk-PBS-0.1% Tween 20. The membranes were then washed three times in 1 X PBS-0.1% Tween 20 and incubated with HRP-conjugated secondary antibodies diluted 1: 2,000 in 5% skim milk-PBS 0.1% Tween 20 for 1 hour. After a successive wash, the membranes were developed using an ECL kit.
6) Determination of cytokine secretion
The cytokine levels in culture supernatants were determined using a commercially available ELISA kit for IL-6 and TNF-α (R&D system, Minneapolis, MN, USA) in accordance to the manufacturer’s instructions.
7) IκB kinase (IKK) assay
Cell extracts for the kinase assay were prepared in a buffer consisting of 20 mM Tris–Cl (pH 7.6), 150 mM NaCl, 25 mM β-glycerophosphate, 2 mM EDTA, 2mM sodium pyrophosphate, 1 mM sodium orthovanadate, 10% glycerol, 1 mM DTT, and 1% Triton X-100. After centrifugation of the lysate at 16,000 xg for 10 minutes at 4°C, the supernatant was incubated with anti-IKKα Ab, which was diluted 1:100, and with 50 ml of protein-G Sepharose beads with endoverend rotation overnight at 4°C. The beads were then washed twice sequentially in buffer A (1 M NaCl, 20 mM Tris-HCl [pH 7.4] and 0.1% Nonidet P-40), buffer B (200 mM NaCl, 20 mM Tris-HCl [pH 7.4], 1% Nonidet P-40, 0.1% SDS and 1 mM EDTA), and buffer C (20 mM Tris-HCl [pH 7.4] and 0.1% Nonidet P-40). Kinase reactions were initiated by the addition of 10 mL of buffer (20 mM HEPES [pH 7.6], 20 mM β -glycerophosphate, 0.1 mM sodium orthovanadate, 10 mM MgCl2, 50 mM NaCl and 1 mM DTT) containing 0.5 mg GST-IκBα (containing aa1–317), and 10 mCi of [g-32P] ATP. The reaction mixture was incubated at 30°C for 30 minutes. The kinase reaction was terminated by adding a protein sample buffer. Kinase reaction products were subjected to SDS/PAGE in 10% gels, followed by a transfer to a nitrocellulose membrane and autoradiography. This membrane was later used for an immunoblot with anti-IKKα Ab to ensure that equal amounts of kinase were immunoprecipitated.
8) NF-κB transcription factor assay
Translocation activity of NF-κB p65 was determined using a commercially available ELISA kit for NF-κB p65 (Active Motif) in accordance to the manufacturer’s instructions and Western blot analysis.
9) Statistical analysis
The results are presented as the mean values ± SD of two or three separate experiments for each test. The level of statistical significance was determined using one way ANOVA (for multiple groups) or Student’s t test (for comparisons between two groups). A p value of <0.05 was considered to be statistically significant.
Results
1) Effect of Arginine vasopressin (AVP) on LPS-induced IL-6 and TNF-α production in RAW 264.7 cells
We measured IL-6 and TNF-α production in the supernatant of RAW 264.7 cells by ELISA to evaluate the effect of AVP on the production of inflammatory markers. After being stimulated with LPS 1 μg/mL for 6 hours, IL-6 and TNF-α production were increased compared with the control. However, the increase of IL-6 and TNF-α was suppressed by a treatment with AVP 10 nM for 4 hours (Fig. 1).
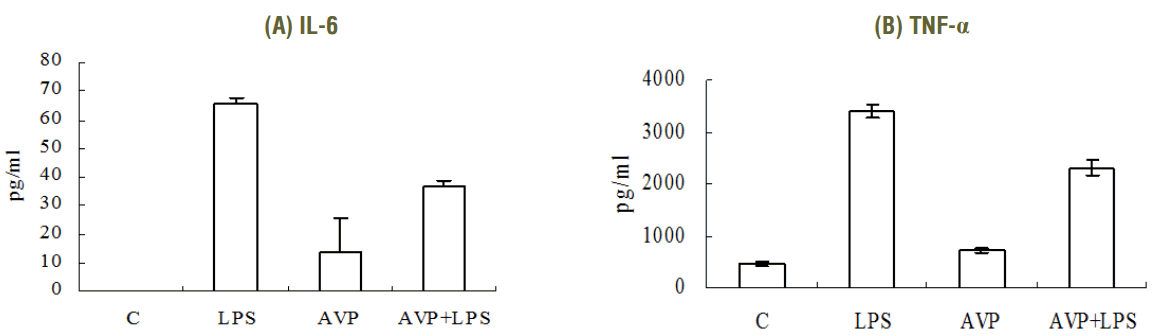
IL-6 and TNF-α expression after vasopressin and/or LPS by ELISA. After stimulation with LPS 1 μg for 6 hours, inflammatory cytokines (IL-6, TNF-α) were increased. However, AVP (10 nM for 4 hours) inhibits inflammatory cytokines production in LPS-induced RAW 264.7 cells compared with the level of LPS alone. The IL-6 level was increased after only AVP applying, but it was not significant (p = 0.199). (A) IL-6, (B) TNF-α. *p < 0.05, compared to the value of LPS alone. IL, interleukin; TNF, tumor necrosis factor, c, control; LPS, lipopolysaccharide; AVP, arginine vasopressin.
2) Effect of AVP on LPS induced IKK activity and IκBa expression in RAW 264.7 cells
To examine the effect of AVP on IKK activity, an immunoprecipitation kinase assay was performed. After a pretreatment with AVP 10 nM for 4 hours and after a treatment with LPS (1 μg/mL) for 5 minutes in AVP-pretreated RAW 264.7 cells, we assessed the IKK activity by phosphorylation of GST-IκBα. The IKK activity was enhanced by LPS and inhibited by AVP (Fig. 2A). Treatment with LPS (1 μg/mL) induced IκBα degradation in RAW 264.7 cells, and maximal degradation was observed after 10 minutes (data not shown). RAW 264.7 cells were pretreated with AVP then stimulated with LPS (1 μg/mL) for 10 minutes. As shown in Fig. 2B, pretreatment with AVP did not have any effect on the amount of baseline IκBα. The amount of IκBα was decreased by treatment with LPS for 10 minutes, and pretreatment with AVP inhibited IκBα degradation.
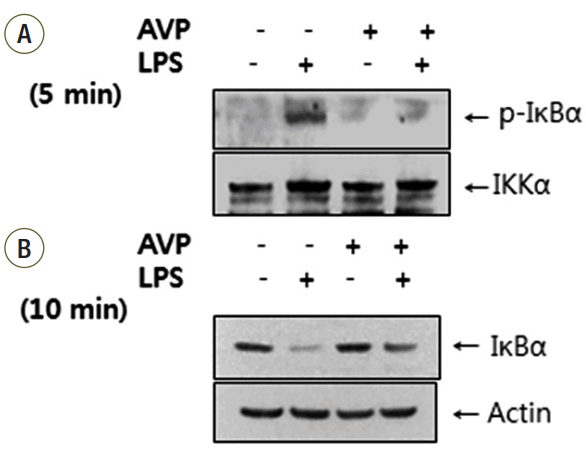
IκB kinase (IKK) activity and IκBα expression after AVP and/or LPS by Western blotting. (A) IKK activity was enhanced by LPS (1 μg/mL for 5 minutes) but pretreated AVP (10 nM for 4 hours) down-regulated IKK activity. (B) IκBα was decreased by treatment with LPS for 10 minutes and pretreatment with AVP blocked IκBα degradation.
3) Effect of AVP on LPS induced nuclear translocation of NF-κB p65 in RAW 264.7 cells
We assessed the effect of AVP on the translocation of p65 subunit of NF-κB from the cytosol to the nucleus after its release from IκBα. AVP pretreatment for 4 hours at 10nM did not affect the amount of cytosolic and nuclear p65. LPS stimulation (1 μg/mL for 30 minutes) alone decreased cytosolic p65 and increased nuclear p65. Otherwise, pretreatment with AVP in LPS-induced RAW cells attenuated NF-κB p65 levels in nuclear fractions and increased NF-κB p65 in cytosolic fractions (Fig. 3).

NF-κB p65 transactivity after AVP and/or LPS by Western blotting. LPS stimulation (1 μg/mL for 30 minutes) alone decreased cytosolic p65 and increased nuclear p65. However, pretreatment with AVP attenuated the NF-κB p65 level changes when LPS was applied in the cytosol and nucleus. A: arginine vasopressin; L: lipopolysaccharide.
4) Effect of AVP on LPS-induced cyclooxygenases-2 (COX-2) production in RAW 264.7 cells
LPS stimulation (1 μg/mL for 8 hours) increased production of COX-2. However, there was no effect of AVP on LPS-induced COX-2 expression (Fig. 4)

Expression of COX-2 after AVP and/or LPS by Western blotting. AVP has no effect on the expression of COX-2. COX: cyclooxygenases.
5) Effect of vasopressin antagonist (tolvaptan) on AVP+LPS induced IκBα expression and nuclear translocation of NF-κB p65 in RAW 264.7 cells
First, we checked whether or not V2R existed in RAW 264.7 cells to confirm the effect of AVP on NF-κB cascade and to evaluate the function of V2 receptor. By using an antibody against V2R, V2R was expressed in a Western blot analysis (data not shown). Then, we treated tolvaptan (V2R antagonist) 100 nM for 4 hours after AVP pretreatment in the cells. As expected, IκBα expression was decreased in LPS-induced RAW 264.7 cells, and pretreatment with AVP inhibited IκBα degradation. When tolvaptan was applied, a decreased IκBα expression was restored (Fig. 5A). The difference of IκBα expression in accordance to tolvaptan dose is shown in Supplementary Fig. 1. Moreover, LPS stimulation increased nuclear translocation of NF-κB p65, and pretreatment with AVP attenuated nuclear translocation. However, nuclear translocation of NF-κB p65 was re-increased in tolvaptan, AVP, and LPS treated cells (Fig. 5B).
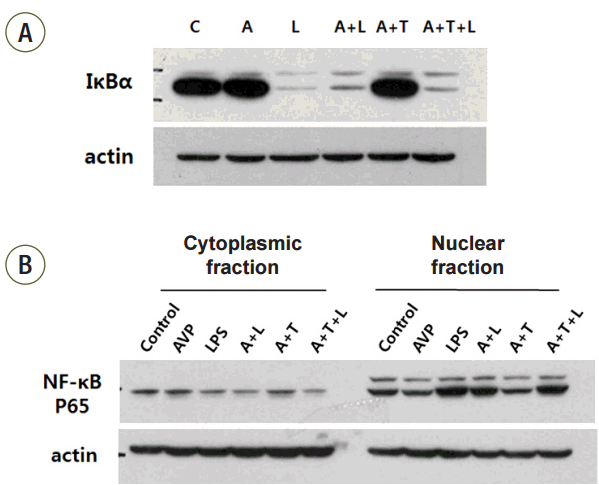
IκBα expression and NF-κB p65 transactivity after AVP and vasopressin receptor antagonist (Tolvaptan) treatment and/or LPS by Western blotting. AVP, tolvaptan, LPS treated cells showed no difference with LPS only stimulated cells on the value of IκBα activation and NF-κB p65 transactivity. (A) IκBα expression. (B) NF-κB p65 transactivity. A, arginine vasopressin; L, lipopolysaccharide; T, Tolvaptan.
Discussion
Our study showed that AVP blocked the effect of LPS stimulation in mouse macrophages. Pretreatment of AVP down-regulated IKK activity, suppressed IκBα degradation, and LPS-induced NF-κB transcription. These results suggest that AVP inhibited nuclear NF-κB binding through the prevention of LPS-induced IκBα degradation. Finally, LPS-induced IL-6 and TNF-α production was decreased by AVP. However, AVP did not affect LPS-induced COX-2 expressions. Therefore, we may conclude that AVP has an anti-inflammatory effect via blocking NF-κB stimulation.
Vasopressin, also known as antidiuretic hormone (ADH), is a nonapeptide synthesized in the hypothalamus. Because the human hormone contains arginine, it is specifically called arginine vasopressin (AVP).[14] AVP and its synthetics are potent alternative vasoconstrictors in the treatment of septic patients with catecholamine-refractive vasodilatory shock.[15] After Landry et al. reported that the AVP plasma levels were inadequately low in patients with vasodilatory septic shock compared to patients with cardiogenic shock,[8] other studies have shown that vasopressin concentrations were elevated in early septic shock, but decreased back to normal range for the majority of patients between 24 and 48 hours as shock continues.[16] As such, many studies regarding AVP in septic shock patients have been carried out. Meanwhile, some studies showed that adrenergic receptor activation down-regulates the pulmonary proinflammatory response in a murine model of sepsis.[11-13] Vasopressin shares some downstream signaling pathways with adrenergic agents; therefore we could hypothesize that vasopressin might modulate an inflammatory response. This study may be meaningful since it shows that AVP may have an anti-inflammatory effect.
In this study, the observed changes in IKK activity, IκBa degradation and NF-κB transcription by AVP was attenuated by tolvaptan treatment. Tolvaptan, an orally active, selective, nonpeptide antagonist that blocks arginine vasopressin from binding to V2 receptors of the distal nephron, often has been used in a variety of water retaining diseases and cardiovascular diseases, including heart failure, hyponatremia, renal diseases, syndrome of inappropriate antidiuretic hormone secretion (SIADH), cirrhosis, and ocular hypertension.[17] The actions of vasopressin are complex due to its action on at least four receptor subtypes: V1 (vascular), V2 (renal), V3 (pituitary), and the OT (oxytocin) receptors.[18] V1R mediates the vasoconstriction, and is expressed mainly in the vascular smooth muscle, cardiac myocytes, and kidney, although less intensely in the kidney; V2Rs are found mainly in the renal collecting duct; and OTRs are thought to mediate tissue specific vasodilation expressed throughout the body.[19] Recently, Chassin et al. reported that vasopressin, via theV2R, reduces inflammation in the renal cell through the inhibition of NF-Κb,[20] and Boyd et al. showed that vasopressin decreased sepsis-induced pulmonary inflammation through V2R in a mouce model.[19] Our study showed similar results with these previous studies.
NF-κB is a family of DNA-binding protein factors, which are required for the transcription of most pro-inflammatory molecules, including certain adhesion molecules (intercellular adhesion molecule 1 [ICAM-1]), critical enzymes (inducible nitric oxide synthase, COX-2), most cytokines (IL-1β, TNF-α, IL-6), and chemokines (IL-8).[6,21,22] Because these molecules are regulated at the level of transcription and are involved in the inflammatory cascade, by inference, NF-κB is a critical intracellular mediator of the inflammatory cascade.[23] NF-κB is a dimer, usually composed of two proteins (p50, p65). In the quiescent cells, NF-κB is sequestered in the cytoplasm by its interaction with a member of the inhibitory IκB family, which includes IκBα and IκBβ. Although a wide variety of stimuli can activate NF-κB, among the most potent inducers are Gram-negative endotoxin or LPS, TNF-α, and IL-1β. After stimulation, IκBα and IκBβ are phosphorylated, polyubiquitinated, and degraded. IκB degradation unmasks nuclear localization peptide sequence signals (NLS) that allow NF-κB to be transported to the cell nucleus, where these dimers are free to bind to DNA containing the sequence that activates gene transcription, including iNOS, COX-2, and pro-inflammatory cytokines.[23,24]
Potential limitation of this study was the use of a single cell line, murine macrophage/monocyte cell line RAW 264.7 cells, obtained from the ATCC. More studies that incorporate other cell lines and multiple strains will be necessary to increase the generalizability of our results.
In conclusion, our results suggest that AVP showed an anti-inflammatory effect on LPS-induced IκBα/NF-κB cascade in mouse macrophages via V2 receptors. A further study about anti-inflammatory effect of AVP in clinical setting is required.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by a Grant (No. 02-2011-050) from the Seoul National University Hospital Research Fund.
Supplementary Materials
The online-only Supplement data is available with this article online: http://dx.doi.org/10.4266/kjccm.2015.30.3.151.