Oxygen therapy for sepsis and prevention of complications
Article information

Abstract
Patients with sepsis have a wide range of respiratory disorders that can be treated with oxygen therapy. Experimental data in animal sepsis models show that oxygen therapy significantly increases survival, while clinical data on the use of different oxygen therapy protocols are ambiguous. Oxygen therapy, especially hyperbaric oxygenation, in patients with sepsis can aggravate existing oxidative stress and contribute to the development of disseminated intravascular coagulation. The purpose of this article is to compare experimental and clinical data on oxygen therapy in animals and humans, to discuss factors that can influence the results of oxygen therapy for sepsis treatment in humans, and to provide some recommendations for reducing oxidative stress and preventing disseminated intravascular coagulation during oxygen therapy.
INTRODUCTION
Sepsis remains a pressing global healthcare concern [1,2]. Moreover, sepsis and septic complications following infection continue to grow worldwide [3,4]. Sepsis and septic shock lead to death in 20%–50% of cases [5,6]. Until the 1990s, the most frequent primary source of sepsis was the abdominal cavity, but in subsequent years, lung infections have become the main reason for the emergence of sepsis [7,8]. Currently, sepsis is most commonly caused by pneumonia; the less frequent causes of sepsis are intra-abdominal infection, primary bacteremia, catheter-inserted infections, urinary tract infections, etc. [9,10]. Moreover, sepsis in pneumonia causes the highest mortality [11]. The bacterial strains that cause sepsis vary over time. Gram-negative organisms have become less common, and gram-positive infections have begun to cause sepsis more frequently [12,13].
Treatment for sepsis includes antibiotics, elimination of infection foci, and maintenance of vital functions and homeostasis [14,15]. Appropriate guidelines, protocols, and clinical recommendations are used for the diagnosis and treatment of sepsis [16-18]. However, they do not contain clear criteria for oxygen therapy [19,20]. At the same time, respiratory disorders in patients with sepsis are overt and have a complex character. The lack of oxygen therapy recommendations seems particularly intriguing given the relatively long history surrounding oxygen in medicine. More than 220 years ago, Thomas Beddoes started to use oxygen for the treatment of asthma, heart disease, and various other illnesses [21], and in 1885, George Holtzapple published the results of using oxygen to treat pneumonia and established its role in the treatment of critical conditions [22].
In sepsis, acute pulmonary injury develops in more than 40% of cases, and acute respiratory distress syndrome develops in 35% of patients [23]. Mortality rate due to the latter complication (approximately 45%) has remained relatively unchanged over the past 40 years [23,24]. Respiratory failure is the main cause of multiple organ dysfunction and failure. In critical conditions, there is a correlation between changes in central venous oxygen saturation (ScvO2; the percentage of oxygen bound to hemoglobin [Hb] in blood returning to the right side of the heart) and the level of tissue hypoxia due to reduced oxygen delivery [23,24]. It is necessary to correct several parameters at once, such as cardiac output, blood Hb count, and arterial blood oxygen saturation, to improve oxygen delivery to tissues. In this case, it is necessary to achieve the target level of 70% ScvO2 [25]. The complexity of correcting many interrelated parameters of gas metabolism in sepsis leads to ambiguous criteria for sepsis oxygen therapy. The purpose of this article is to compare and contrast experimental and clinical data on oxygen therapy in animals and humans, to discuss factors that can influence the success or failure of oxygen therapy for sepsis (OTS) in humans, and to provide some recommendations to reducing oxidative stress and preventing disseminated intravascular coagulation (DIC) in septic patients during oxygen therapy.
HYPERBARIC OTS IN ANIMALS
Experiments in rats and mice provide most of the experimental evidence regarding the efficacy of OTS in animal models. These experiments use different techniques for provoking sepsis, such as cecum ligation and puncture (CLP) and intra-abdominal pathogen injections [26]. Animal experiments are necessary for many reasons, but the extrapolation of experimental data into clinical practice can be improper [27]. Dissimilarities in interactions between pathogen and host, anatomical and physiological differences, variable human translatability of causative pathogenic sepsis agents, and many other factors are obstacles to the application of animal data in the clinic [28,29]. In most experiments, OTC increases animal survival. In rat models with a mortality rate of 100% without treatment, intermittent hyperbaric oxygen therapy (HBOT; breathing oxygen in a pressurized chamber or tube) reduced the mortality rate to 8% (P<0.005); in a model with a mortality rate of 79% without treatment, intermittent HBOT reduced the rate to 23% (P<0.005) [30]. It is easier to experimentally demonstrate the effectiveness of HBOT in different experimental models than to establish the optimal HBOT time [31]. Early HBOT (98% oxygen under pressure up to 2.4 atmospheres within 1 hour after CLP) reduces the systemic inflammatory response [32]. The protective role of HBOT in CLP-induced sepsis may be related to the expression of interleukin (IL)-10 by peritoneal macrophages [33]. Other mechanisms describing the positive action of HBOT are also possible [34].
These mechanisms include the "superinduction" of heme oxygenase-1, inhibition of nitric oxide (NO) production and prevention of lung damage caused by lipopolysaccharides, reduction of inflammatory mediators due to HBOT, modulation of nuclear factor-kappa B activity and inhibition of excessive myeloperoxidase production, suppression of bacterial growth in the small intestine, preservation of erythrocyte deformability, and acceleration of free radical acceptor synthesis to mitigate the effects of reactive oxygen species (ROS). Routine use of HBOT in addition to antibiotic therapy may increase survival in sepsis [35]. In addition, ozone therapy is highly effective in increasing animal survival after intraperitoneal pathogen inoculation [36]. HBOT and ozone therapy reduce oxidative stress and myeloperoxidase synthesis, decrease the concentration of tumor necrosis factor (TNF)-α and IL-1β, and prevent lung damage [36]. In CLP-induced sepsis, the use of 98% O2 (2.4 atmospheres) provides a 52% survival rate compared to 13% in the control group [32]. The positive effect is represented by a decrease in the expression of TNF-α, IL-6, and IL-10 [32,37]. Thus, most experimental data demonstrates the beneficial effects of oxygen therapy in the treatment of sepsis.
OTS IN HUMANS
Analysis of the data available from Medline, the Cochrane Database of Systematic Reviews, and other sources indicated that different aspects of HBOT optimal use for sepsis treatment need further research [38]. More research is also needed regarding the use of normobaric oxygen (adjuvant oxygenation by nasal cannula or facemask at one atmosphere of pressure), which has the potential to be as effective as the use of antibiotics [39]. Normobaric oxygen should be used immediately after the development of sepsis; otherwise, it will not increase patient survival [40]. At the same time, other aspects of OTS are also important and should also be studied [41]. Data on the partial pressure of oxygen (PaO2) in arterial blood indicate that both high and low PaO2 can increase sepsis mortality [42]. Even though hyperoxia stimulates the production of ROS, HBOT is used to treat sepsis in humans [43-45]. Oxygen therapy reduces mortality by stimulating the anti-inflammatory response and suppressing the pro-inflammatory response [30,46]. Although early HBOT has a positive effect on anti-inflammatory parameters and mortality, it is not known whether delayed oxygen therapy prevents the development of septic shock [47,48]. Delayed HBOT promotes the oxidation of succinate to fumarate in the mitochondria and prevents the accumulation of succinate, which leads to organ damage in sepsis [49]. Although theoretical explanations for the benefits of OTC are numerous, the adequate practical use of oxygen in sepsis does not have precise criteria An individualized approach is necessary to decrease the risk of oxygen injury to the lungs. Oxygenation of 90% and tolerable hypercapnia are considered acceptable [50].
In contrast, liberal oxygen therapy induces hyperoxemia in most patients and decreases their survival. Mild hyperoxemia decreases oxygen delivery [51]. High oxygen levels in the central venous blood (ScvO2, 88%–100%) increase mortality in patients with sepsis [52]. High oxygen supplementation (FiO2>65%) and hypoxemia (PaO2<7 kPa) can increase in-hospital mortality [53]. Short-term hyperoxia (<7–10 minutes) can cause atelectasis [54]. Providing normoxemia may reduce the negative effects of hyperoxemia in sepsis [55]. HBOT can provoke a depletion of leukocytes and platelets and cause DIC [56]. Although liberal oxygen therapy has been a cornerstone of therapy for sepsis, alertness in hyperoxia necessitates titration of OTS to normoxia [57]. Hyperoxia also increases mortality in patients who achieve normoxia after hospitalization and therefore requires targeting for normoxia after the initiation of mechanical ventilation, which may improve patient outcomes [58]. Hyperoxia can cause damage to the alveoli, pulmonary edema, and a systemic inflammatory response; therefore, targets should be chosen to reduce oxygenation [59,60]. Hyperoxia also changes the composition of surfactant proteins; suppresses mucociliary clearance; causes vasoconstriction; and decreases coronary blood flow, cardiac output, and microvascular perfusion [61]. On the other hand, until now, the idea of “acceptable hypoxemia” has not been tested [62]. But if not corrected, tissue hypoxia can cause multiple organ dysfunction and death. As a result, OTS is performed in many patients even without documented hypoxia. Hypoxia significantly increases mortality, which increases in parallel with PO2 [63].
Thus, OTS may worsen some situations rather than improve survival [42,64]. Oxygen is a medicine: it has both the desired positive effects and dangerous side effects. The balance between the positive and negative effects of higher and lower doses of OTS is still unknown [65-67].
OXIDATIVE STRESS IN SEPSIS AND OXYGEN THERAPY
Given that oxygen is a drug, its narrow range of therapeutic efficacy and safety in sepsis should be recognized. The toxic effects of hyperoxia are well known; particularly, high barometric pressure of oxygen (more than two atmospheres) “poisons” the brain, causing the Paul Ebert effect: nausea, blurred vision, coughing, trouble breathing, headache, dizziness, muscle twitching, irritability, disorientation, and convulsions among other effects [68-70]. Although oxygen therapy is recommended for many specific pathologies and clinical situations, for example, necrotizing fasciitis, chronic wounds, diabetic retinopathy, and carbon monoxide poisoning [71-74], the use of oxygen for sepsis requires a cautious approach. Bacteremia is one of the reasons that increased caution should be taken surrounding the use of oxygen in sepsis. Bacteremia complicates OTS because it increases oxidative stress in the blood plasma. The increase in oxidative stress is associated with a mechanism for removing bacterial infection from the bloodstream in humans, the main mechanism for doing so being oxycytosis [75]. This occurs in arterial blood, where red blood cells kill bacteria through the oxygen released by oxyhemoglobin. Usually, the amount of oxygen released is limited by the amount required to kill bacteria, and oxycytosis does not lead to significant oxidation of blood plasma or a decrease in its redox potential [76].
The situation changes with sepsis. The latter is primarily caused by the so-called sepsis-causing bacteria that can survive oxidation. Being resistant to active oxygen and ROS, they provoke red blood cells to release the maximal possible amount of oxygen from oxyhemoglobin (Figure 1). The released oxygen dissolves in the blood plasma. Oxygen therapy, especially HBOT, raises the quantity of dissolved oxygen in the blood plasma [58-61]. The increased concentration of dissolved oxygen causes oxidation of plasma components (proteins, lipids, hormones, amino acids, peptides, vitamins, etc.),depletion of the antioxidant capacity of plasma, and platelet activation [56,76]. Oxidation of plasma components causes disruption of hormonal regulation and homeostasis [77]. Depletion of the antioxidant capacity of plasma causes damage to cell membranes and DNA [78]. Oxygen dissolved in blood plasma activates platelets, which can cause DIC after activation by oxygen [79-82]. Activated platelets also generate ROS [83] following glycoprotein (GP) IIb/IIIa receptor stimulation, arachidonic acid metabolism, glutathione (GSH) cycle, phosphoinositide metabolism, etc [84,85]. As a result, ROS generation continues as a chain reaction. Sepsis-causing bacteria penetrate the erythrocyte membranes and cause Hb leakage and hemolysis [77,79]. In turn, intravascular hemolysis activates platelets due to direct activation by Hb, increased ROS, nitrogen oxide (NO) removed by liberated Hb, and the release of intra-erythrocyte adenosine diphosphate [86]. Platelets are also activated by extracellular neutrophil traps, histones, and aggregates of platelets and leukocytes [87-89]. Thrombin also activates platelets. With sepsis, an overproduction of thrombin occurs, which increases inflammation. Thrombin activates platelets via GPIbα and protease-activated receptors PAR1 and PAR4 [90]. Thrombin generation occurs through the fibrin network, extracellular neutrophil traps, and histones [91]; expression of encoded tissue factor, cytokines, and microparticles by endothelial cells [92]; initiation of bacterial polyphosphate via the “contact” pathway [93]; and invasion into endothelium by bacteria and, as a result, platelet activation (via FcγRIIa, αIIbβ3, and platelet factor-4) [94]. During infection, thrombin generation can be stimulated by many other pathways as well [95,96].

Oxidation of plasma components, platelet activation and depletion of the antioxidant capacity of blood plasma by oxycytosis. ROS: reactive oxygen species; DIC: disseminated intravascular coagulation.
Thus, the mechanism of bacteria-killing in the bloodstream by oxygen released from oxyhemoglobin (oxycytosis) demonstrates one of the risks of using oxygen in the treatment of sepsis. Excessive release of oxygen from erythrocytes, together with the use of OTS, can significantly increase the amount of oxygen dissolved in blood plasma. Dissolved oxygen causes many harmful effects, and one of the most serious complications is platelet activation, which can initiate DIC. Although oxycytosis and OTS alone can cause DIC. Together, they do so more quickly and easily. HBOT for sepsis dissolves more oxygen in the blood than normobaric OTS. Therefore, the use of HBOT in sepsis is more dangerous than that of normobaric OTS.
HENRY’S LAW AND OXYGEN THERAPY IN SEPSIS
HBOT can have unexpected effects on oxygen transport. It is known that blood carries oxygen in two forms: dissolved in plasma and bound to Hb. Dissolved oxygen is not involved in oxygen transfer due to its low solubility. Arterial blood with 100 mm Hg PO2 dissolves only 0.3 ml of O2 per 100 ml of blood, while 100ml of Hb contains 20 ml of oxygen [97]. Dissolved oxygen takes part in oxygen transport when 100% oxygen is inhaled, which typically raises alveolar PO2 to over 600 mmHg and increases the concentration of dissolved oxygen from 0.3 to 2 ml in 100 ml of blood. This amount of dissolved oxygen is 40% of the difference in arterial-venous oxygen concentrations of 5 ml O2/100 ml of blood [98]. Oxygen dissolves in blood according to Henry's law [99,100]. The law says that the quantity of O2 dissolved in blood is proportional to PO2:[O2]=k PO2, where k = (0.003 ml O2/100 ml of blood)–1 mm Hg–1 (Figure 2). The oxygen concentration in arterial blood correlates with the partial pressure of inhaled O2, the intensity of ventilation and gas exchange, the level of Hb, and the affinity of Hb for O2 [101]. At a high partial pressure of inhaled O2, the role of dissolved oxygen in O2 transfer increases, and the role of Hb decreases [102]. At a PaO2 of more than 2.2 atmospheres, Hb ceases to transport oxygen to the tissues, and as a result, tissue respiration is provided by oxygen dissolved in the blood plasma [103]. If the PaO2 for breathing is three atmospheres, the blood dissolves about 6 vol% oxygen (6 ml of O2 in 100 ml of plasma), and this amount of oxygen corresponds to the normal oxygen consumption of the human body (arteriovenous oxygen difference) when erythrocytes pass through the capillaries without emitting oxygen and not binding carbon dioxide [102-104]. As a result, a paradoxical situation may develop: the more oxygen the patient breathes under pressure, the less oxygen is delivered to the tissues and the less carbon dioxide is removed from the tissues (Figure 3).
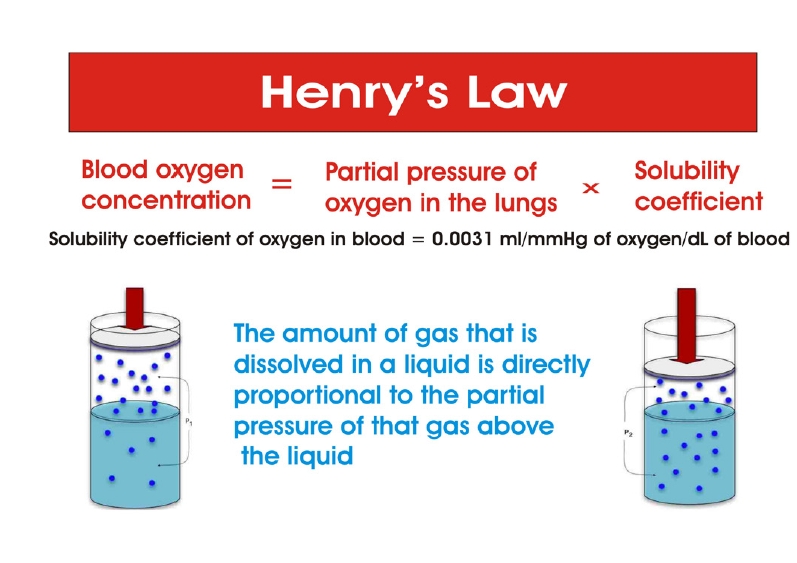
Henry's law and the dissolution of oxygen in blood plasma.

Gas transport by erythrocytes in normal physiology (A) and hyperbaric oxygen therapy (B).
Many publications show that hyperoxia increases sepsis mortality [52-67]. It is difficult to recommend a specific oxygen therapy policy covering all clinical situations, so OTS should be used with caution. In patients with sepsis, along with clinical, instrumental, and laboratory parameters, plasma oxidative stress and blood clotting should also be monitored.
MARKERS OF OXIDATIVE STRESS AND PLATELET ACTIVATION
In sepsis, the markers of oxidative stress indirectly show the intensity of oxygen release by erythrocytes during oxycytosis and the amount of dissolved oxygen in the blood plasma. Oxidative stress can be detected by finding oxidants, such as free radicals; testing the level of antioxidants; measuring oxidation products, including protein carbonyl, F2-isoprostanes, malondialdehyde, and 8-oxo-20-deoxyguanosine (8-OHdG); and measuring the redox balance check (ratio of reduced GSH/oxidized GSH). It is reasonable to test different oxidative markers simultaneously [105].
In clinical practice, oxidative stress is often detected by separate testing of human serum albumin, plasma proteins, vitamin C, etc. Serum albumin is the main antioxidant against ROS [106]. It contains a cysteine residue (Cys34) that is oxidized by the formation of a disulfide bond with free cysteine amino acids [107]. Oxidative damage to proteins increases the level of carbonylation [108]. This biomarker is formed early and is stable [109]. Carbonyl groups are formed in various ways, for example, by cleavage of the main protein chain, in particular at proline, lysine, and arginine side chains, , and by lysine deamination [110]. The most commonly used methods for testing carbonyl groups are enzyme-linked immunosorbent assay and high-performance liquid chromatography HPLC. They are popular due to their high throughput and standardization [111].
The antioxidant system includes superoxide dismutase, GSH peroxidase, catalase, and non-enzymatic antioxidants (ascorbic acid, tocopherols, bilirubin, GSH, etc.). Ascorbic acid (vitamin C) is useful for the evaluation of oxidative stress. It scavenges free radicals, hydroxyl radicals, hydrogen peroxide (H2O2), singlet oxygen, and others [112,113]. For the assessment of oxidative stress in vivo, new reliable and simple tests have been proposed and adapted for automatic analyzers. These tests allow for fast processing of many samples and avoid manual handling of samples and reagents.
Given that oxidative stress activates platelets and, thus, can trigger DIC [56,76], antiplatelet therapy for sepsis is imperative. It is necessary to constantly check the condition of blood coagulation, with particular attention to tests for platelet function, for adequate antiplatelet therapy. Numerous tests show platelet function. Monitoring of antiplatelet therapy and diagnosis of platelet dysfunction are necessary to assess the risk of DIC and bleeding [114]. Moreover, markers of platelet activation can help predict sepsis [115].
Born’s test, developed in the 1960s, was the first test to diagnose platelet function. Aggregometry of light transmission in platelet-rich plasma reveals platelet aggregation in response to various agents causing aggregation in vitro [116]. In the 1980s, other tests of platelet function entered clinical practice, such as ex vivo flow cytometry, measurement of nucleotides and platelet-specific compounds, aggregometry of whole blood platelets, etc. [117]. Platelets are also easily activated during blood sampling and blood manipulation [118]. New, simpler, and more reliable platelet function tests are currently being developed. They provide testing of platelet function directly at the point of care or at the patient's bedside and are also becoming available in general laboratories, hospitals, and in everyday practice [119]. Currently, various assays are used to monitor antiplatelet therapy, including the VerifyNow P2Y12 assay, Multiplate analyzer, VASP-P (vasodilator-stimulated phosphoprotein-phosphorylation), PFA-100/200 (platelet function analyzer), and others. They overcame the problems of previous tests, such as Born's aggregometry. Monitoring antiplatelet therapy in OST is necessary to make adequate therapeutic decisions and control the treatment of sepsis [119].
It is important to be aware of the risks of OTS and to predict possible complications before they begin. Some preventive measures to reduce the risk of DIC may be prudent. Before and during oxygen therapy, certain medications may be required to increase the antioxidant potential of blood plasma and to prevent platelet activation and aggregation.
NEUTRALIZATION OF ROS IN PLASMA AND INCREASING THE ANTIOXIDANT POTENTIAL OF PLASMA
Innate immune mechanisms for clearing the bloodstream of bacterial infection include oxycytosis - the destruction of bacteria by oxygen released from erythrocytes [75-77]. The bacteria that cause sepsis are resistant to oxygen, and red blood cells release the maximum amount of oxygen. Excess oxygen depletes the antioxidant capacity of plasma and causes redox imbalances, in particular, a decrease in GSH levels. GSH provides protection against oxidative stress. It is synthesized from cysteine, glycine, and glutamate [105]. A decrease in plasma GSH activity increases sepsis mortality [106,107]. In plasma, GSH not only traps ROS but also removes toxins and drugs [108]. GSH is also depleted as sepsis progresses to septic shock [109]. To prevent DIC, septic shock, and multiple organ failure (MOF), GSH should be included in sepsis therapy as soon as infection enters the bloodstream and oxycytosis begins. There are currently no tests to detect the early stages of bacteremia and oxycytosis, and GSH therapy should be started in conjunction with antibiotic therapy as soon as sepsis is suspected [105-107]. The chemical structure of synthetic GSH is similar to that of natural GSH produced in the human body, so overdose is rare. GSH is non-toxic; it has very few side effects and interacts weakly with some drugs, such as tetracycline, sulfanilamide, and vitamin B12. Intramuscular injections of GSH (600 mg in 4 ml of 0.9% NaCl) are possible, but intravenous infusions of GSH are preferred. The total dose of GSH depends on the degree of plasma redox potential depletion [106-108]. GSH can be used with several antioxidant vitamins, such as vitamins C, E, and A.
Vitamin C (ascorbic acid) can be used to increase the redox potential of plasma alone or together with GSH and vitamins A and E. As an antioxidant vitamin, ascorbic acid is preferred over vitamin E and vitamin A due to its water solubility and low toxicity. Humans cannot synthesize vitamin C, while other animals rapidly increase their production during oxidative stress [120]. Vitamin C is a neuroprotector, immunomodulator, and cofactor for the synthesis of vasopressors [121]. High doses of vitamin C have been used for many years in complementary and alternative medicine [122]. Vitamin C has a protective effect against oxidative stress in sepsis and septic shock [123]. High doses of vitamin C reduce the degree of multiple organ failure [124]. In a double-blind, randomized clinical trial, 28-day mortality was decreased following vitamin C administration [125,126]. Vitamin C in combination with thiamine and hydrocortisone also significantly reduces sepsis mortality [122]. Some clinicians began using ascorbic acid in their daily practice for the treatment of sepsis before testing for ascorbic acid in clinical trials [127]. Early studies used 1 to 2 g intravenous doses of ascorbic acid every 8 hours. The high dose of ascorbic acid (1,584 mg/kg/day) was also well tolerated [126]. The antioxidant effect of ascorbic acid is dose-dependent and is maximal at plasma concentrations >175 mg/L [128]. Optimal doses for intravenous infusion are approximately 10 g per day but may be higher [126-128]. High plasma concentrations of ascorbic acid can only be achieved by intravenous infusion, not oral administration [127].
High doses of vitamin C can cause mild renal impairment and glucose-6-phosphate dehydrogenase deficiency. Calcium oxalate nephropathy is a rare, potentially toxic effect of vitamin C. Side effects are dose-dependent, but no adverse clinical reactions have been reported in most patients and healthy volunteers [121-123]. It is possible for ascorbic acid to become a standard component of sepsis therapy as more data are uncovered [128].
There are conflicting publications regarding the use of retinol (vitamin A), tocopherols (vitamin E), and vitamin D in the treatment of sepsis and septic shock [129-138]. The use of these vitamins for sepsis requires further study. Again, it should be noted that antioxidant therapy for the prevention of DIC should be carried out before platelet activation by oxygen and ROS; otherwise, therapy may be less effective or even ineffective.
PREVENTION OF PLATELET AGGREGATION
Neutralizing oxygen and ROS in plasma and increasing plasma redox potential can prevent platelet activation that causes DIC. However, if antioxidant therapy fails and platelet activation occurs, early anticoagulant therapy is indispensable [139]. The state of coagulation must be quickly and repeatedly assessed to identify the initiation of DIC syndrome [140]. Particular attention should be paid to platelets [141,142]. Intensive anticoagulant therapy reduces in-hospital mortality [143]. Antiplatelet medications prevent DIC and reduce mortality or complications in critically ill patients [144-146]. Antiplatelet therapy before hospitalization and early targeted therapy reduces the risk of DIC [147]. In experimental models of sepsis, aspirin or P2Y12 inhibitors prevent organ failure and mortality without increasing bleeding [148]. Platelet formation can be inhibited either by aspirin (acetylsalicylic acid), which suppresses COX-1activity and blocks the synthesis of thromboxane A2, or by clopidogrel and ticagrelor, which stop platelet activation induced by adenosine diphosphate [149]. Aspirin is a cheap and relatively safe drug at low doses for platelet inhibition that can effectively reduce mortality from sepsis [150,151]. Aspirin can also be used in low-grade DIC treatment [152]. Low-dose aspirin decreases the need for intensive care [153,154]. A large study of 7,945 intensive care unit (ICU) patients showed the effectiveness of aspirin for increasing sepsis patient survival [155]. Another study analyzed 886 patients with sepsis and found that aspirin significantly reduced mortality in ICUs and hospitals [156]. On the other hand, a recent study has shown that daily low-dose aspirin treatment did not reduce deaths associated with sepsis in community-dwelling older adults [157].
Clopidogrel and ticagrelor may be alternatives to aspirin to prevent DIC. These antiplatelet drugs may become a new approach to the prevention of DIC and multiple organ failure [158,159]. Ticagrelor inhibits platelet aggregation faster and more efficiently than clopidogrel [160]. The mortality risk is lower with ticagrelor therapy compared with clopidogrel therapy [161]. Ticagrelor also inhibits the reuptake of adenosine. Clopidogrel and other thienopyridines do not affect adenosine metabolism [162].
Heparin can also be used to prevent DIC; it is not an antiplatelet agent. The anticoagulant effect of heparin is determined by the activation of antithrombin III, which inactivates thrombin, activated factor X (factor Xa), and other proteases [163]. Heparin can reduce DIC and MOF [164]. It decreases mortality in sepsis [165]. Improvement of the state of hypercoagulability in sepsis with heparin reduces the incidence of DIC or multiple organ failure and also reduces overall ICU stay [166]. Heparin decreases mortality in sepsis without causing bleeding [167].
DISCUSSION
The results of oxygen therapy in the experimental treatment of sepsis in animal models contradict the results of the use of oxygen in clinical practice. HBOT in experimental sepsis increases survival, reduces oxidative stress, decreases the concentration of TNF-α and IL-1β, prevents lung damage, and has other positive effects by suppressing the expression of TNF-α, IL-6, and IL-10 [20-27]. In contrast to experimental data, clinical data on the use of oxygen in patients with sepsis are not exactly definitive both regarding hyperbaric and normobaric oxygen. When using normobaric oxygen as part of sepsis treatment, both an increase and a decrease in the mortality rate were recorded [30,40,42,46]. Data on the use of hyperbaric oxygen in sepsis are also inconsistent. Early HBOT may have a positive effect on anti-inflammatory parameters and mortality [47,48], while delayed HBOT can prevent organ damage from sepsis [49].
However, the majority of clinical studies [51-64] demonstrate that HBOT can increase mortality by causing decreased oxygen delivery to tissues; alveolar damage, pulmonary edema, and atelectasis; vasoconstriction, decreased coronary blood flow, cardiac output, and microvascular perfusion; and DIC. Perhaps the negative effects of hyperbaric oxygenation in sepsis treatment can be partially explained by Henry's law, the presence of additional oxidative stress in sepsis, and a decrease in the redox potential of plasma due to oxycytosis. Thus, neutralizing ROS in plasma and increasing the antioxidant potential of plasma to mitigate some of the negative effects of HBOT through the use of GSH, ascorbic acid, and other antioxidants may be reasonable. As for DIC syndrome, which can be triggered by sepsis and HBOT, early anticoagulant therapy and platelet inactivation seem to be indispensable.
The results of the studies carried out to date on the use of oxygen in the complex therapy of sepsis are contradictory and do not assist the development of effective and safe protocols for the use of oxygen in sepsis treatment; on the other hand, the continuation of research and the further accumulation of statistical data in the form in which it happened before will hardly help the development of adequate protocols for oxygen therapy with respect to sepsis treatment. The only thing that can be more or less confidently asserted from the data accumulated to date is that normobaric OTS is safer than HBOT. An individualized approach to OTS has many nuances, requires a physician's personal experience, and can be difficult to implement in modern conditions of treating patients by a group of doctors. Perhaps new innovative basic theoretical and clinical research is needed to provide a breakthrough in the optimization of OTS and to ensure the development of appropriate optimal protocols.
CONCLUSION
Sepsis causes a wide range of respiratory disorders that require oxygen therapy. The use of oxygen therapy in animal models of sepsis is effective and improves animal survival in the majority of experiments. The use of different protocols for oxygen therapy in patients with sepsis gives less unequivocal results. Oxygen therapy, especially HBOT, increases the concentration of dissolved oxygen in plasma, depletes plasma antioxidant potential, suppresses oxygen transport by erythrocytes, decreases carbon dioxide removal from the tissues, activates platelets, and increases the risk of thrombosis and DIC. Oxygen therapy should be performed under the control of plasma oxidative stress tests. Oxidative stress should be corrected by changing the parameters of oxygen therapy and the use of antioxidants (GSH, vitamins C, A, and E). During oxygen therapy, the blood coagulation state should be constantly tested, paying special attention to platelet function tests. If necessary, anticoagulants, such as aspirin, clopidogrel, ticagrelor, and heparin, should be used after sepsis diagnostics and during oxygen therapy.
Notes
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.